A Swiss Army Knife of Methods
Deciphering the structure, dynamics and interactions of non-canonical DNA

ARNA, INSERM U1212, CNRS UMR 5320, Université de Bordeaux
UFR des Sciences Pharmaceutiques, Université de Bordeaux
May 12, 2026
Hello from Bordeaux



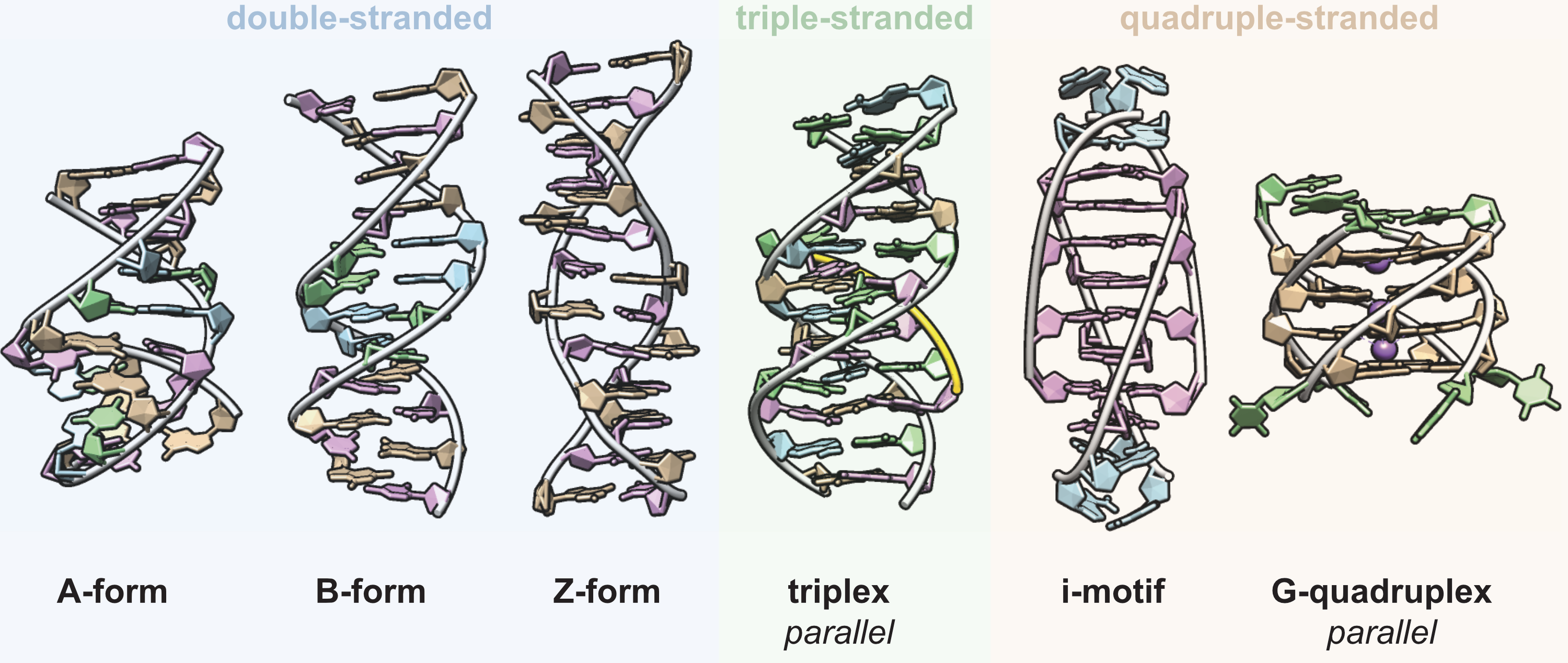
DNA can adopt many secondary structures
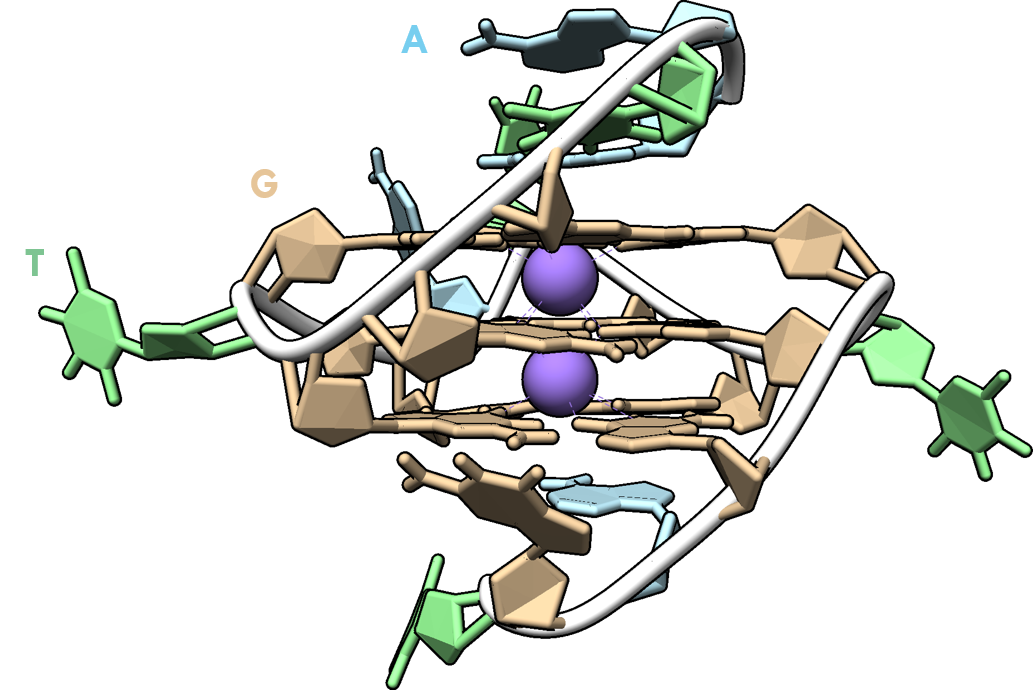
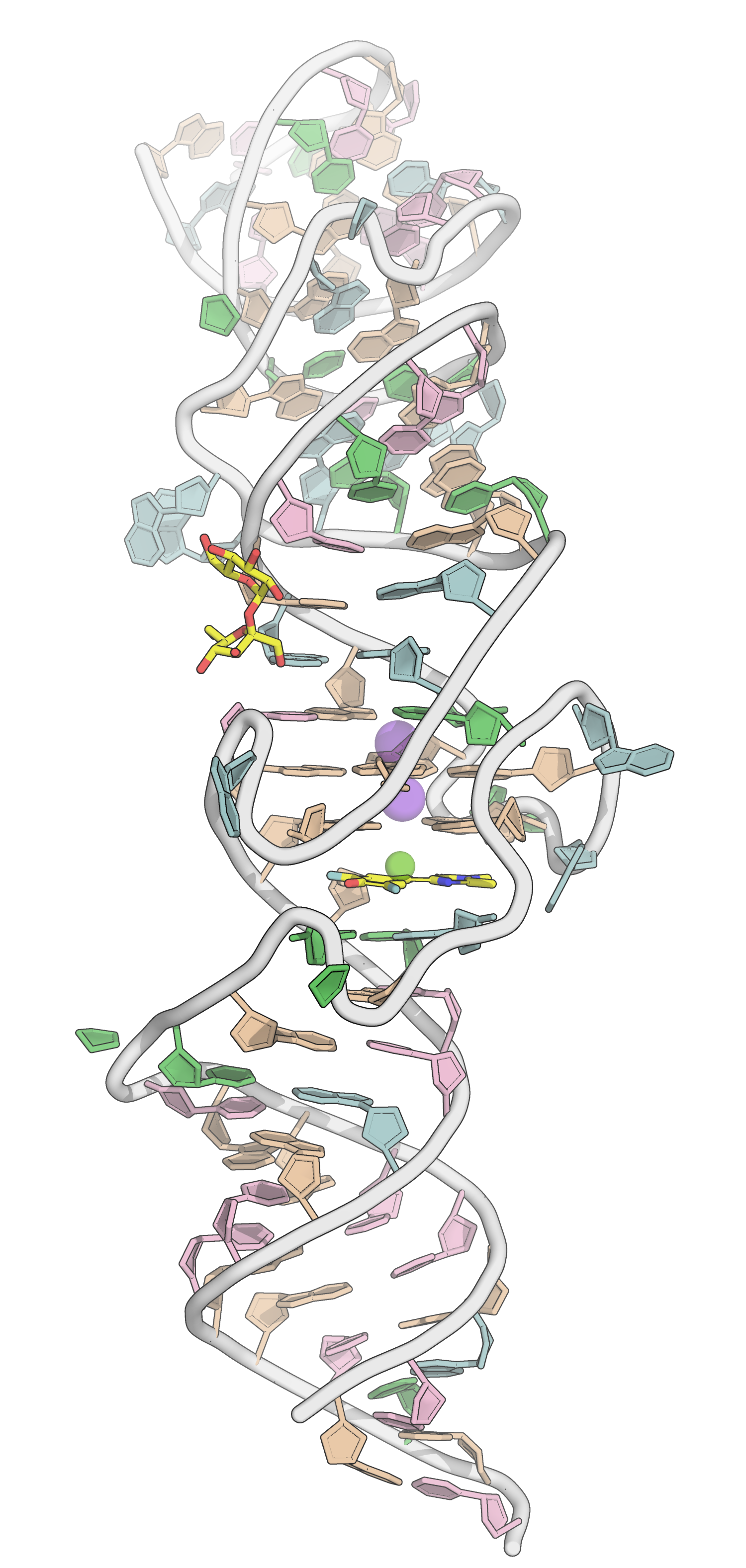
G-quadruplexes are particularly peculiar
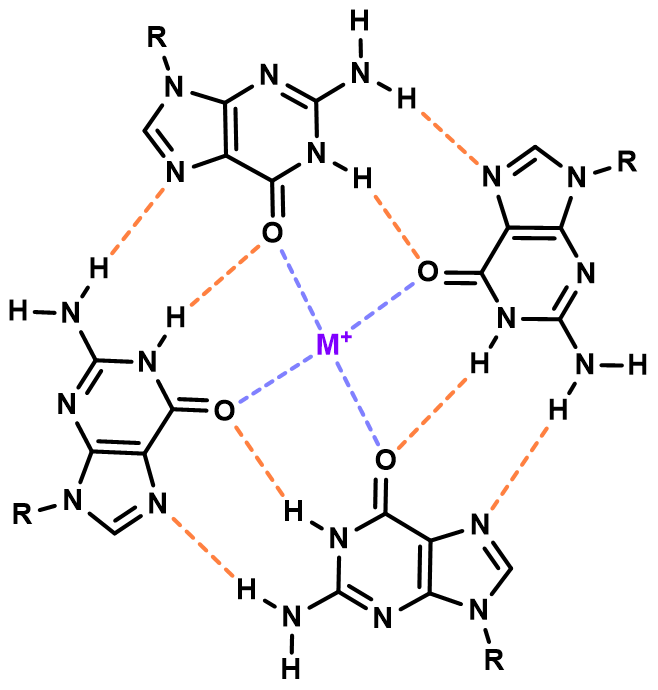
G-quadruplexes are particularly peculiar
G4s are DNA/RNA drug targets distinct from dsDNA
G4s may also be drugs themselves
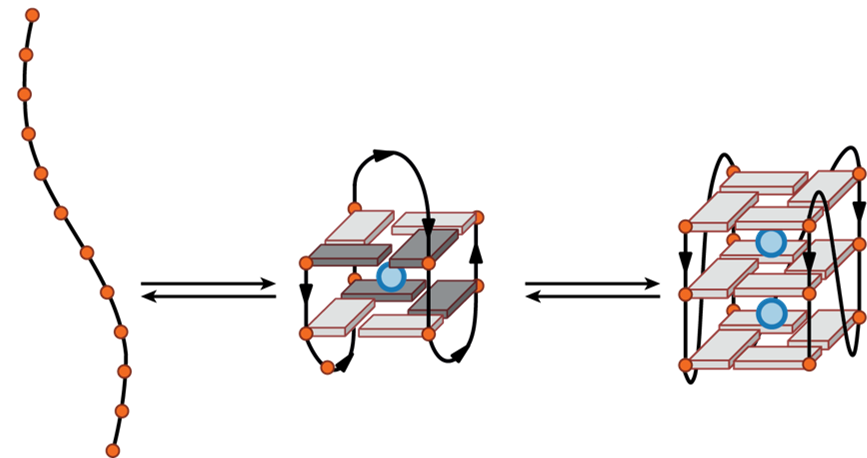
G4s are very polymorphic…
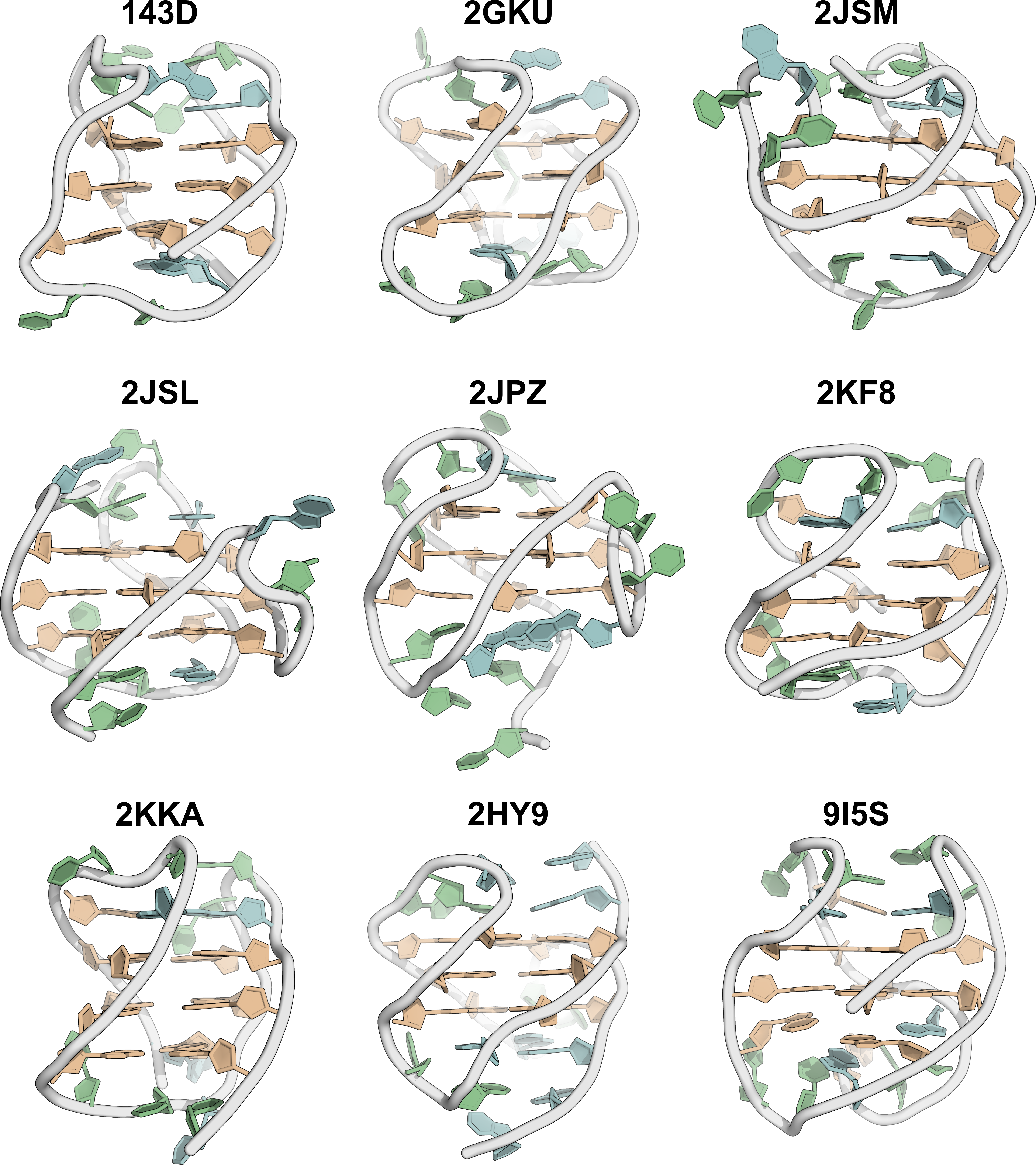
…and in equilibrium
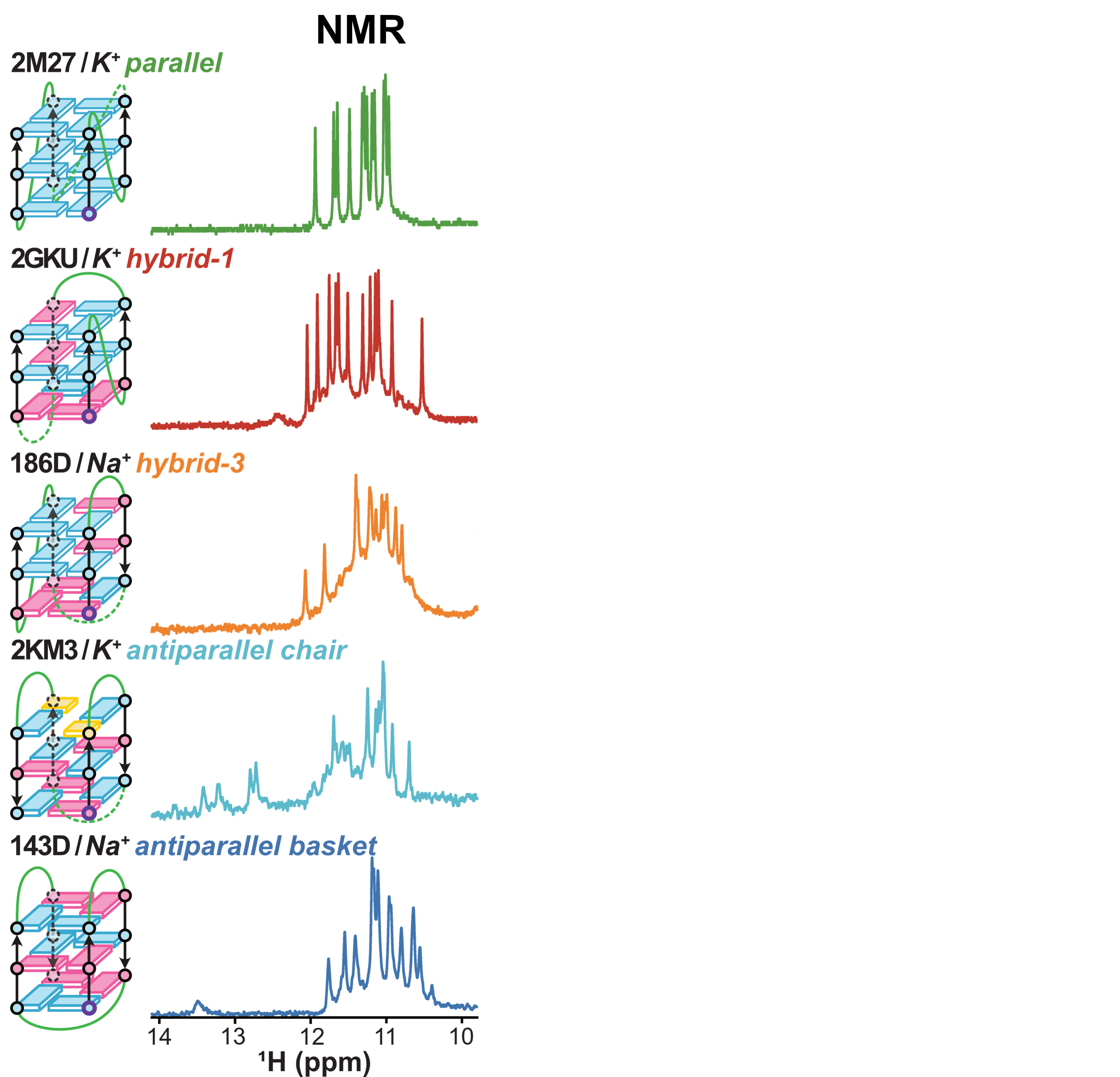
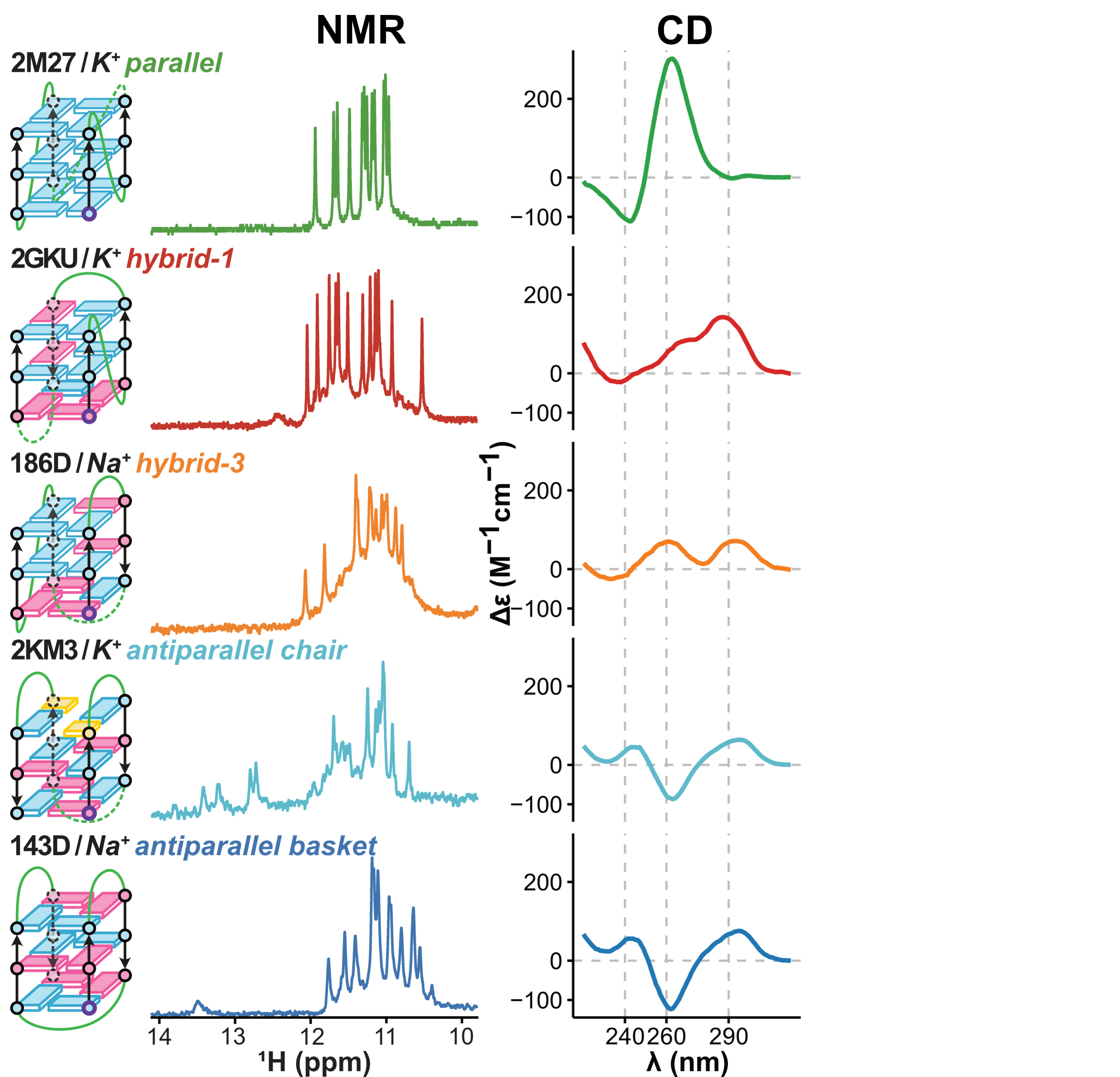
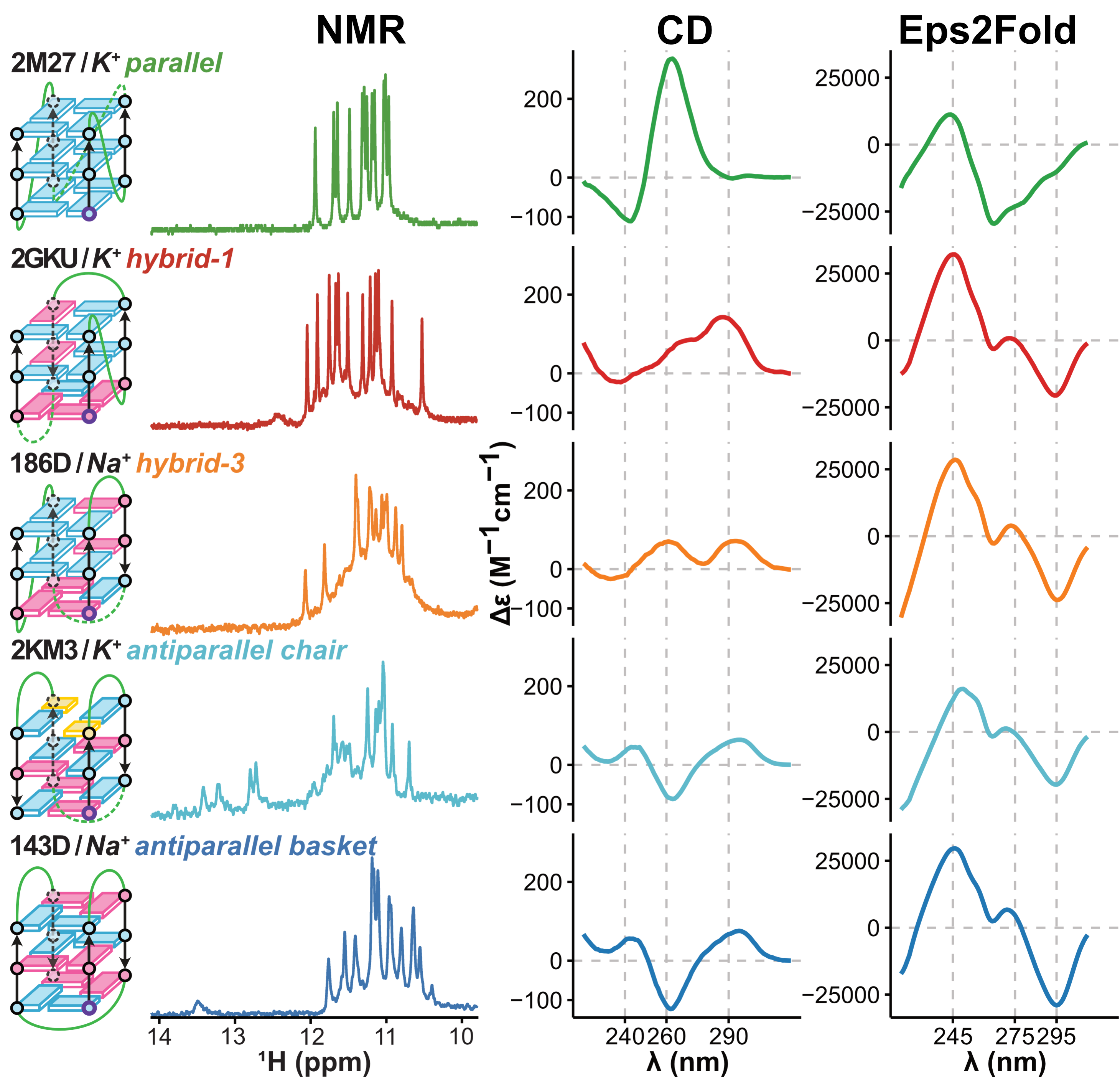
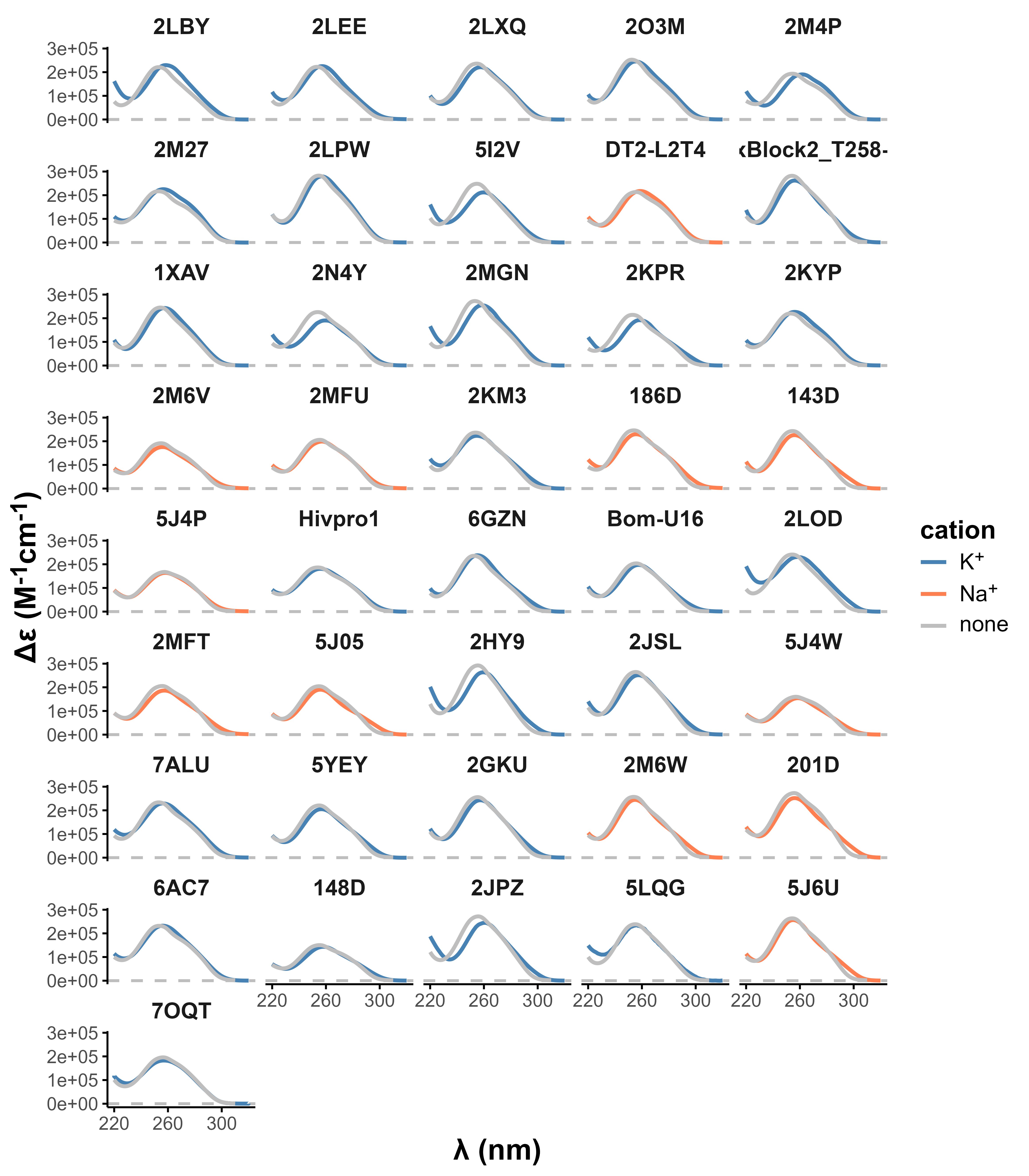
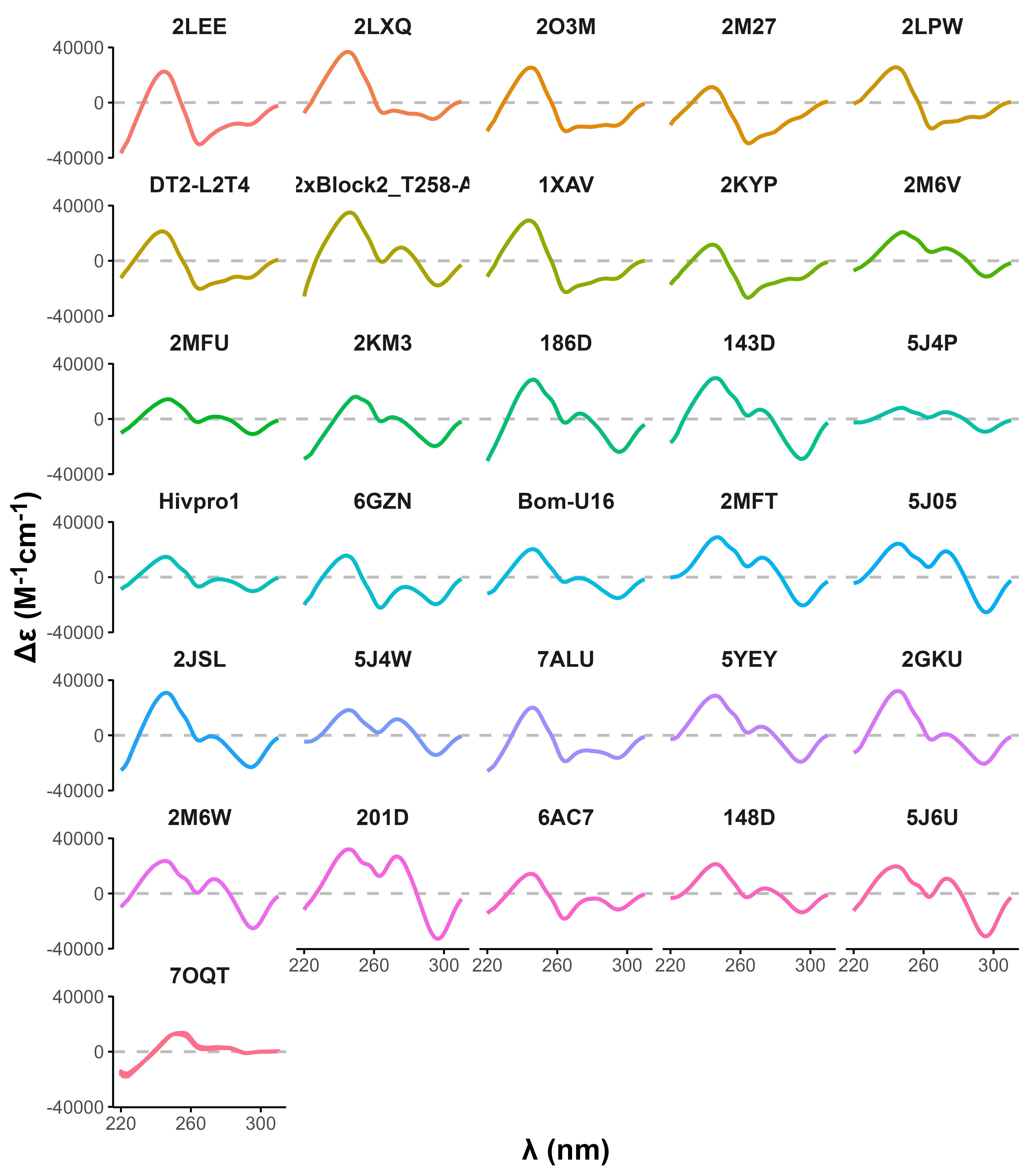
Topologies are poor structural descriptors
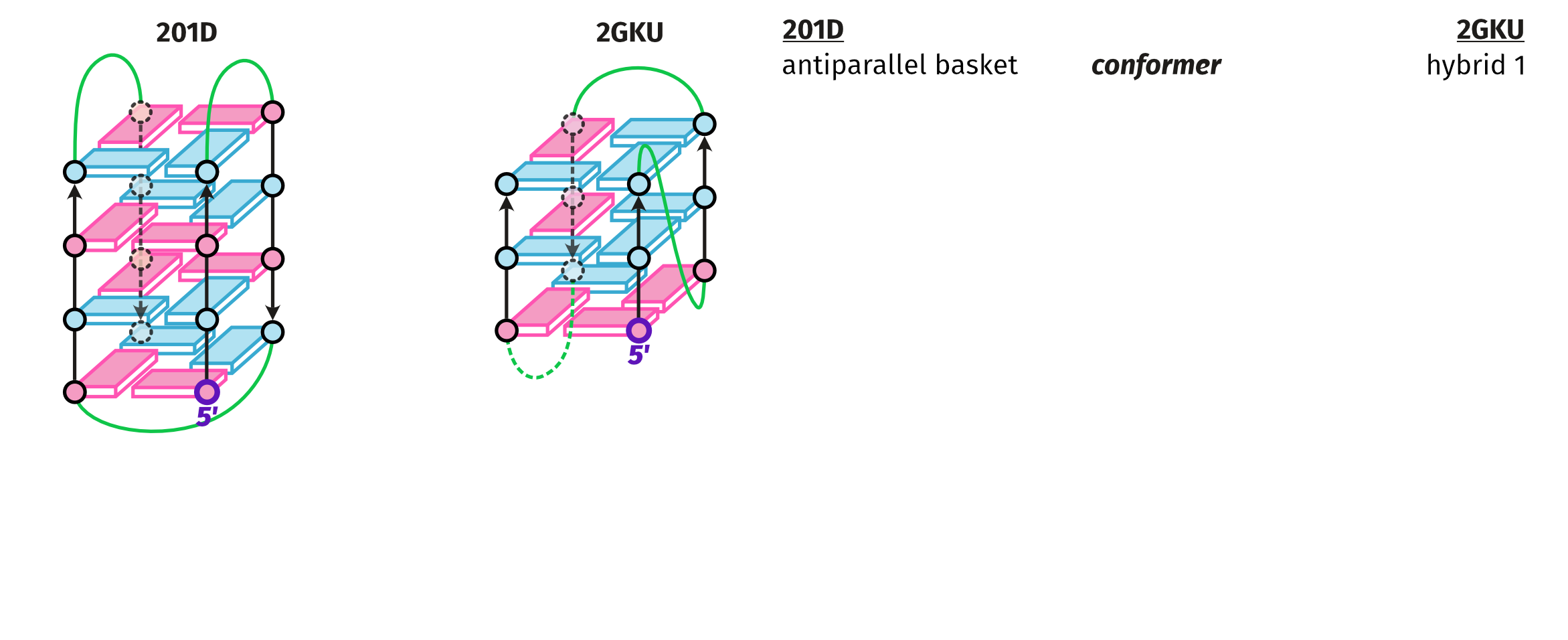
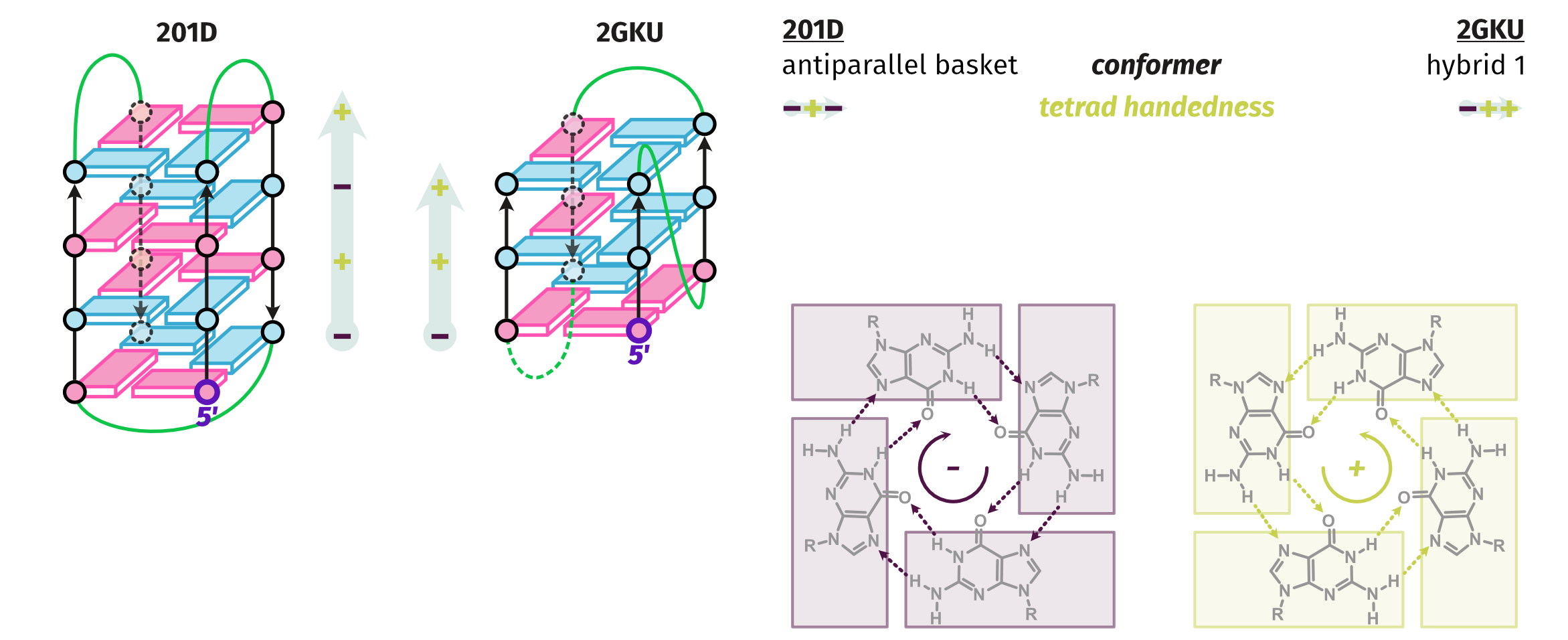
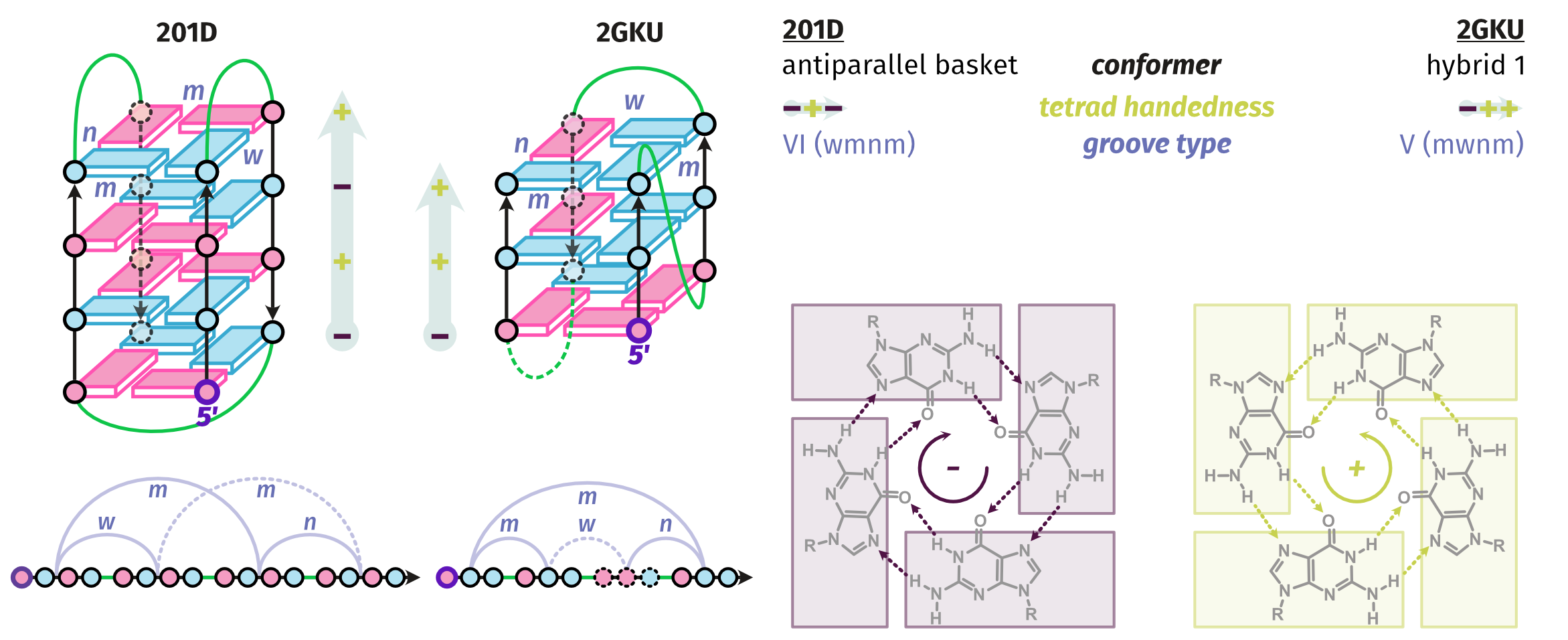
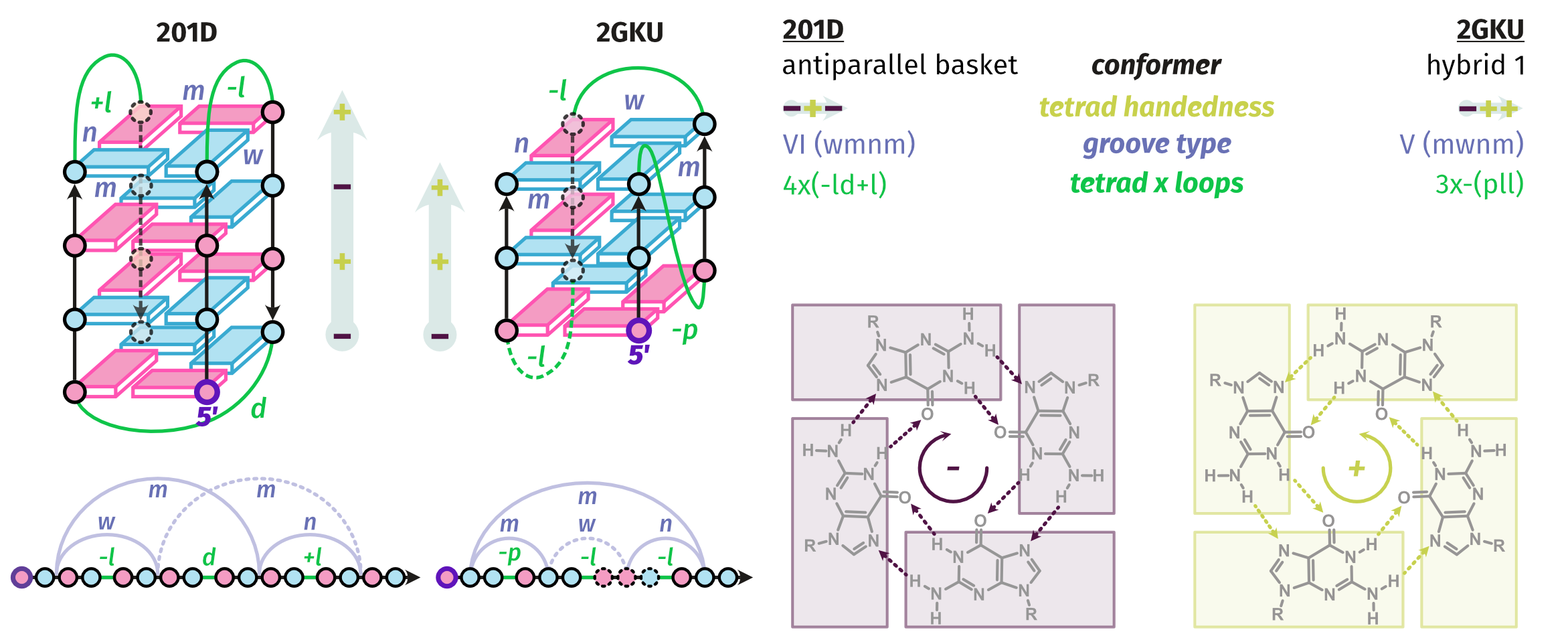
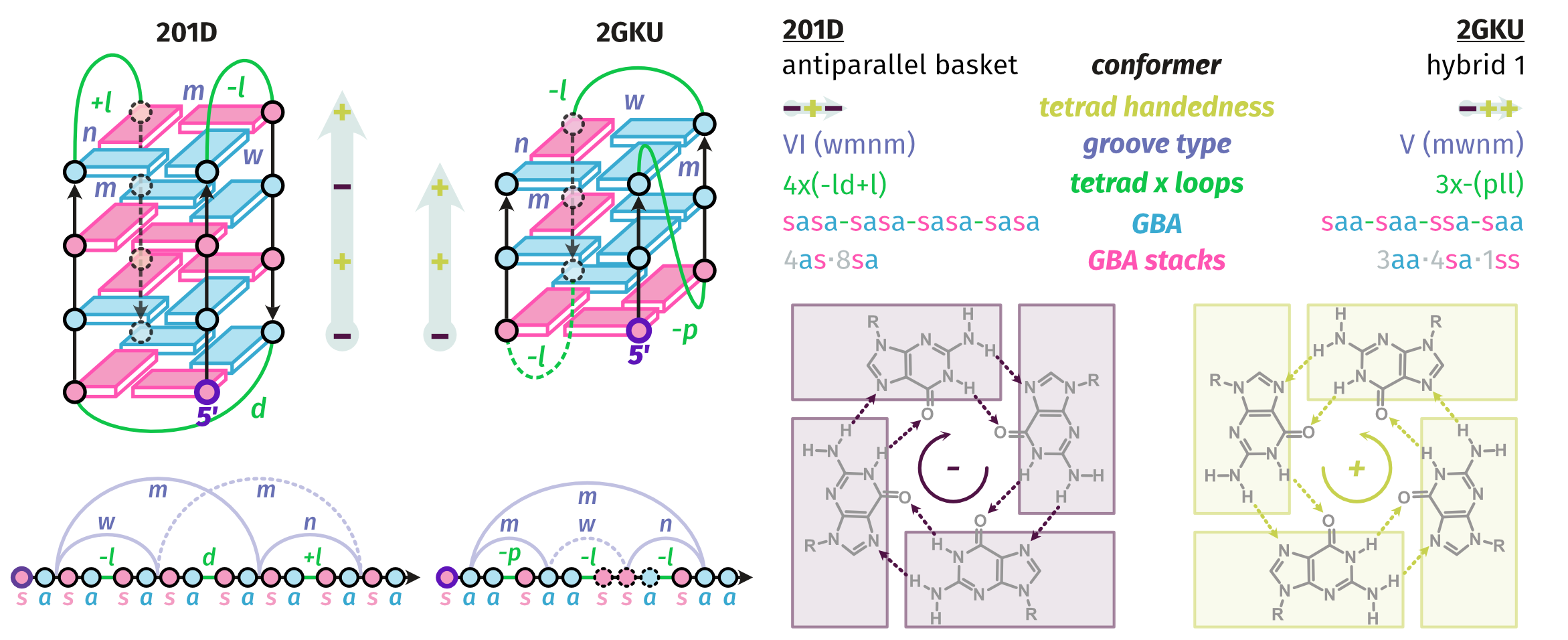


Topologies are poor structural descriptors


Stability measurements are often innacurate
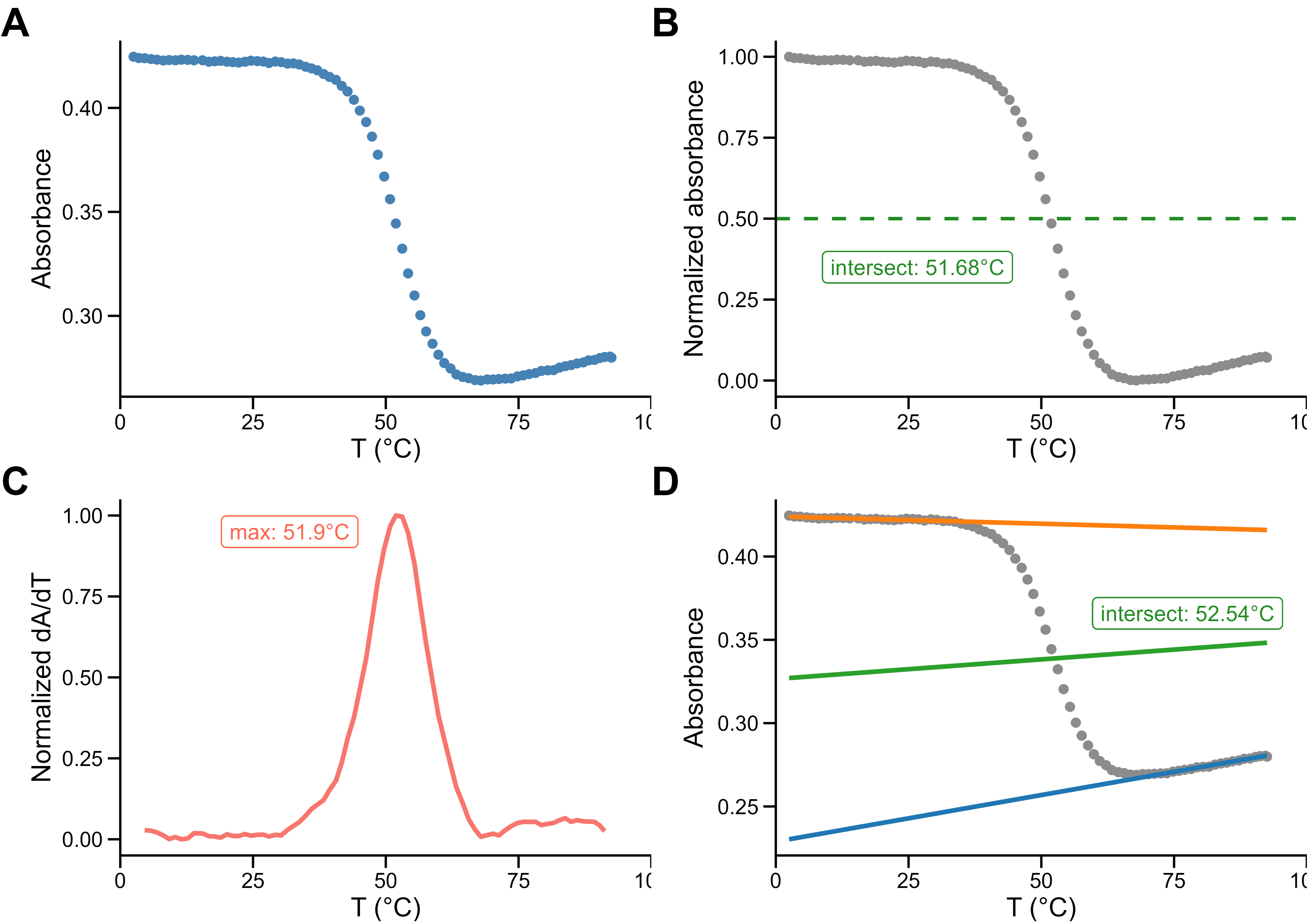
Stability measurements are often innacurate
\[ A_T=(a^FT+b^F) \times \frac{1}{1+exp(-\frac{\Delta H^0 (1- \frac{T}{T_m})}{RT})} + (a^UT+b^U) \times \frac{exp(-\frac{\Delta H^0 (1- \frac{T}{T_m})}{RT})}{1+exp(-\frac{\Delta H^0 (1- \frac{T}{T_m})}{RT})} \]
\[ A_T=\textcolor{forestgreen}{(a^FT+b^F)} \times \frac{1}{1+exp(-\frac{\Delta H^0 (1- \frac{T}{T_m})}{RT})} + \textcolor{forestgreen}{(a^UT+b^U)} \times \frac{exp(-\frac{\Delta H^0 (1- \frac{T}{T_m})}{RT})}{1+exp(-\frac{\Delta H^0 (1- \frac{T}{T_m})}{RT})} \]
\[ A_T=(a^FT+b^F) \times \frac{1}{1+exp(-\frac{\Delta H^0 (1- \frac{T}{\textcolor{coral}{T_m}})}{RT})} + (a^UT+b^U) \times \frac{exp(-\frac{\Delta H^0 (1- \frac{T}{\textcolor{coral}{T_m}})}{RT})}{1+exp(-\frac{\Delta H^0 (1- \frac{T}{\textcolor{coral}{T_m}})}{RT})} \]
\[ A_T=(a^FT+b^F) \times \frac{1}{1+exp(-\frac{\textcolor{steelblue}{\Delta H^0} (1- \frac{T}{T_m})}{RT})} + (a^UT+b^U) \times \frac{exp(-\frac{\textcolor{steelblue}{\Delta H^0} (1- \frac{T}{T_m})}{RT})}{1+exp(-\frac{\textcolor{steelblue}{\Delta H^0} (1- \frac{T}{T_m})}{RT})} \]
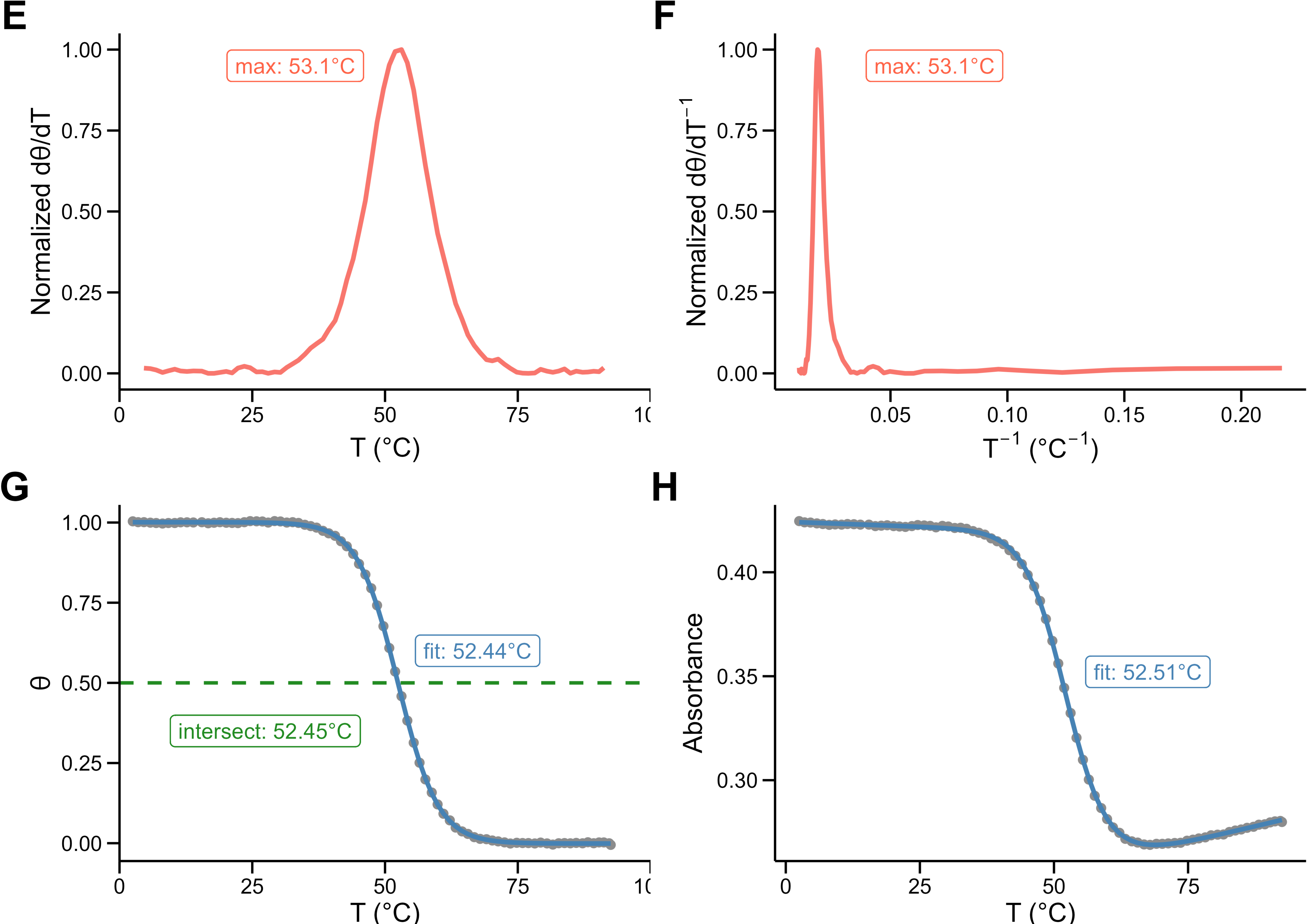
meltR allows high-throughput and robust Tm determination
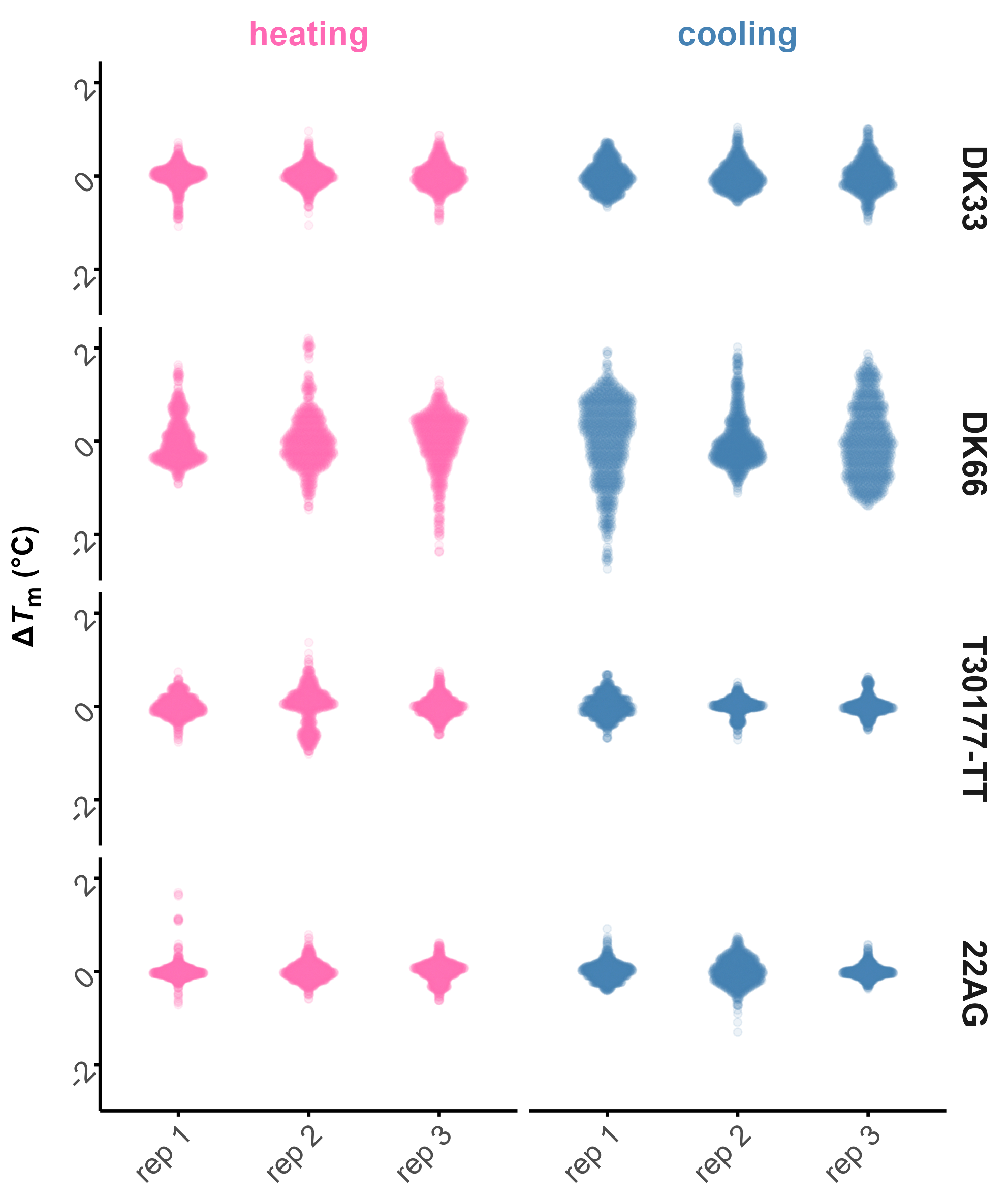
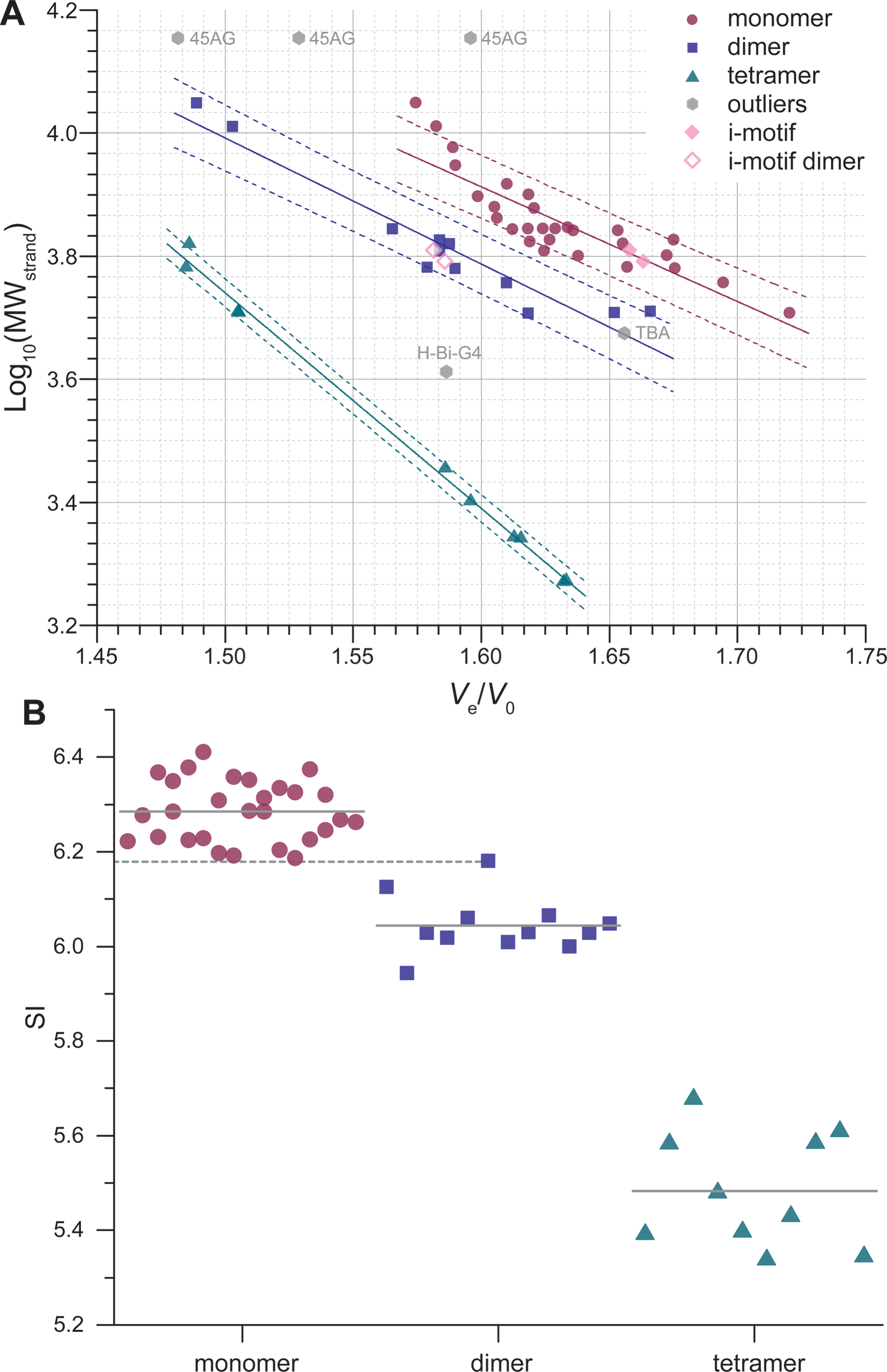
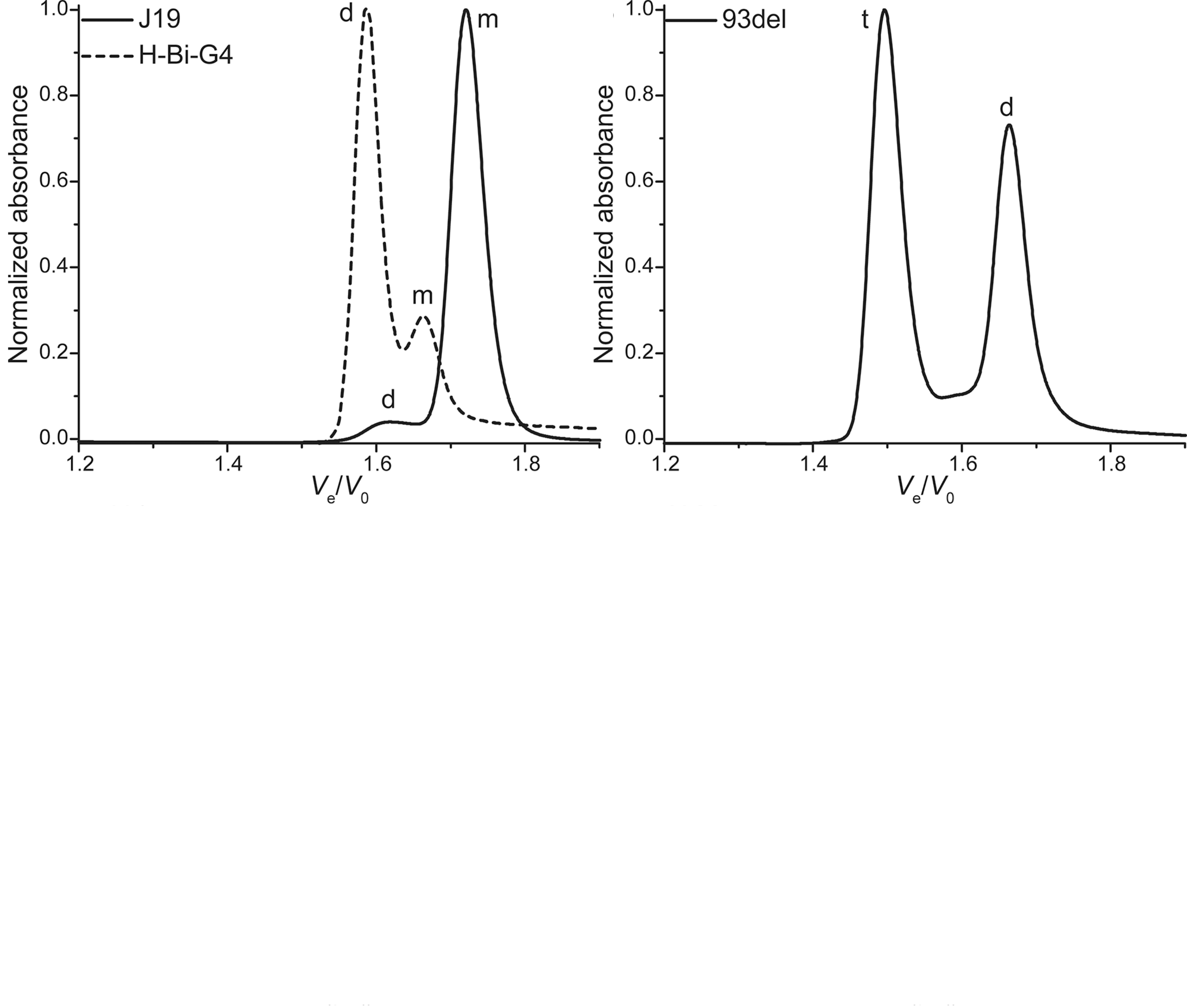
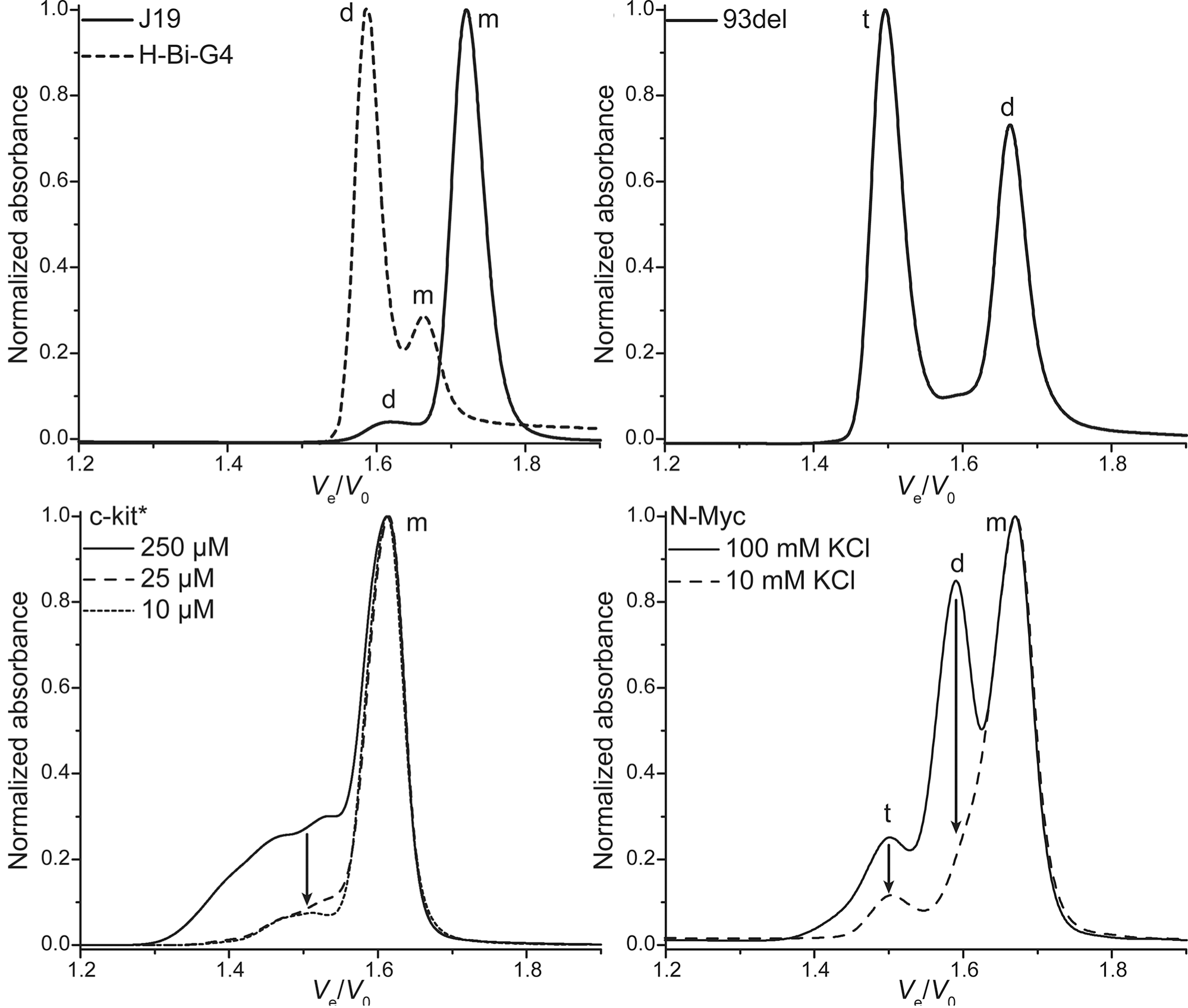
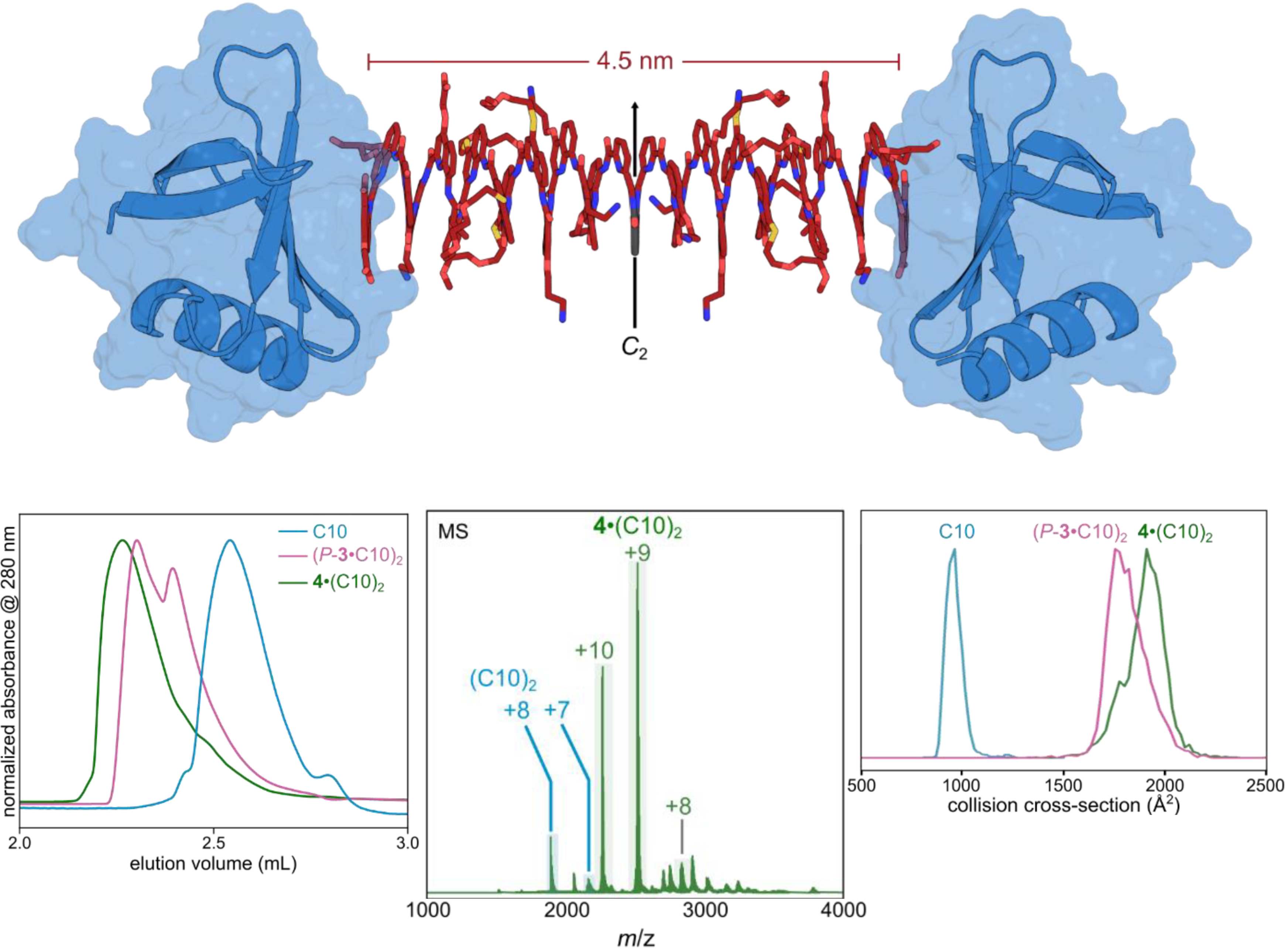
SEC allows robust stoichiometry determination
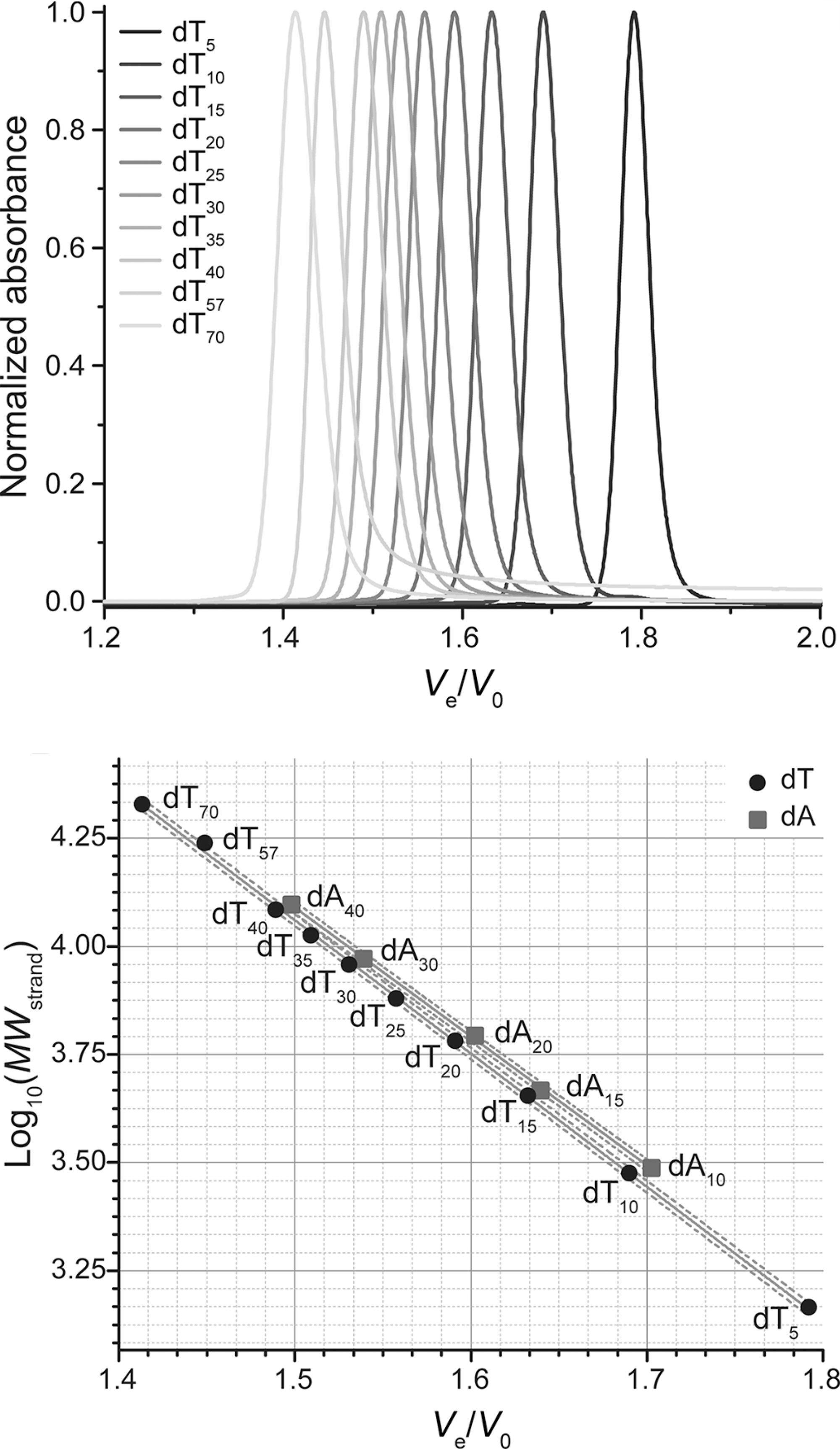
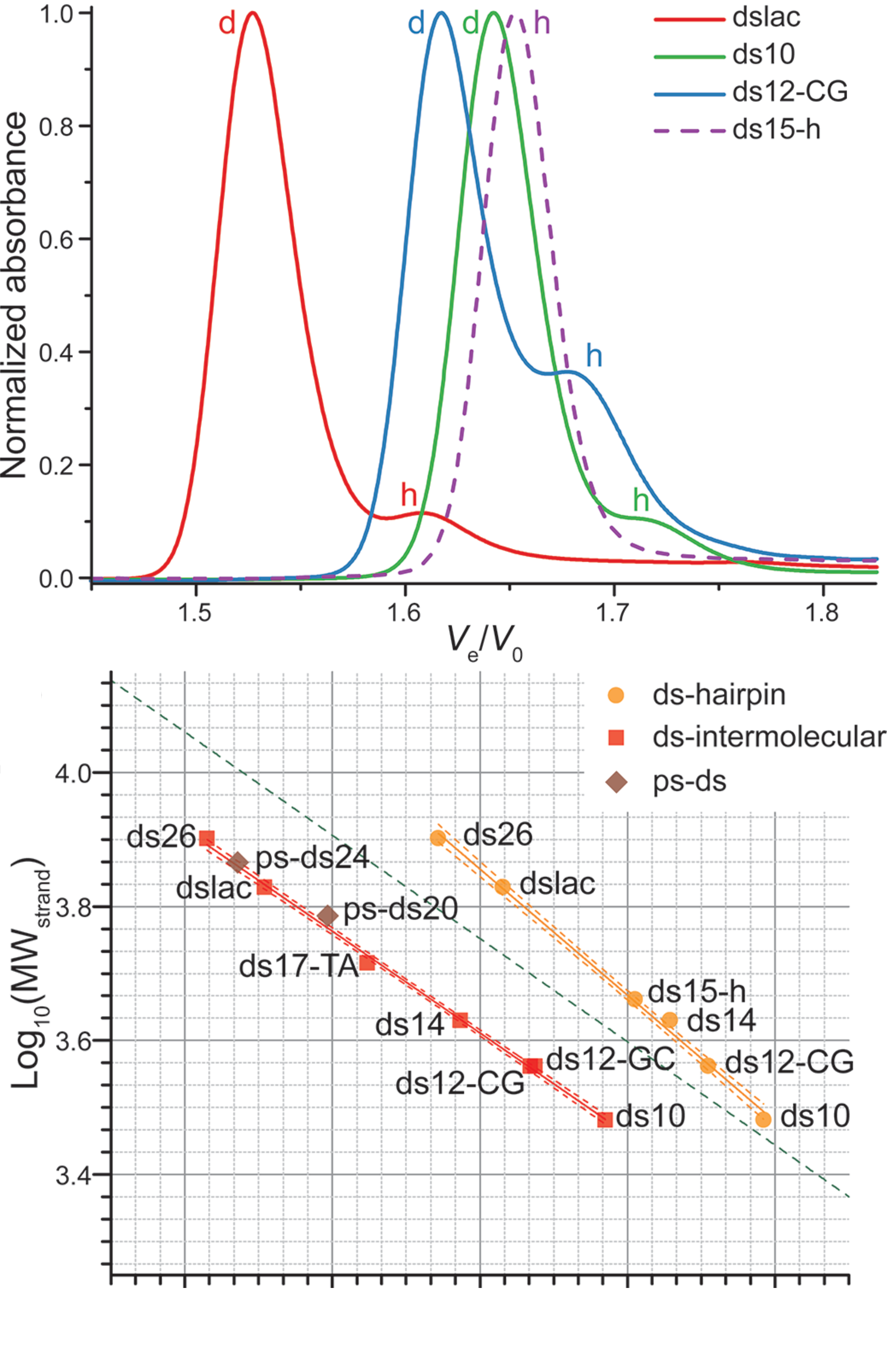
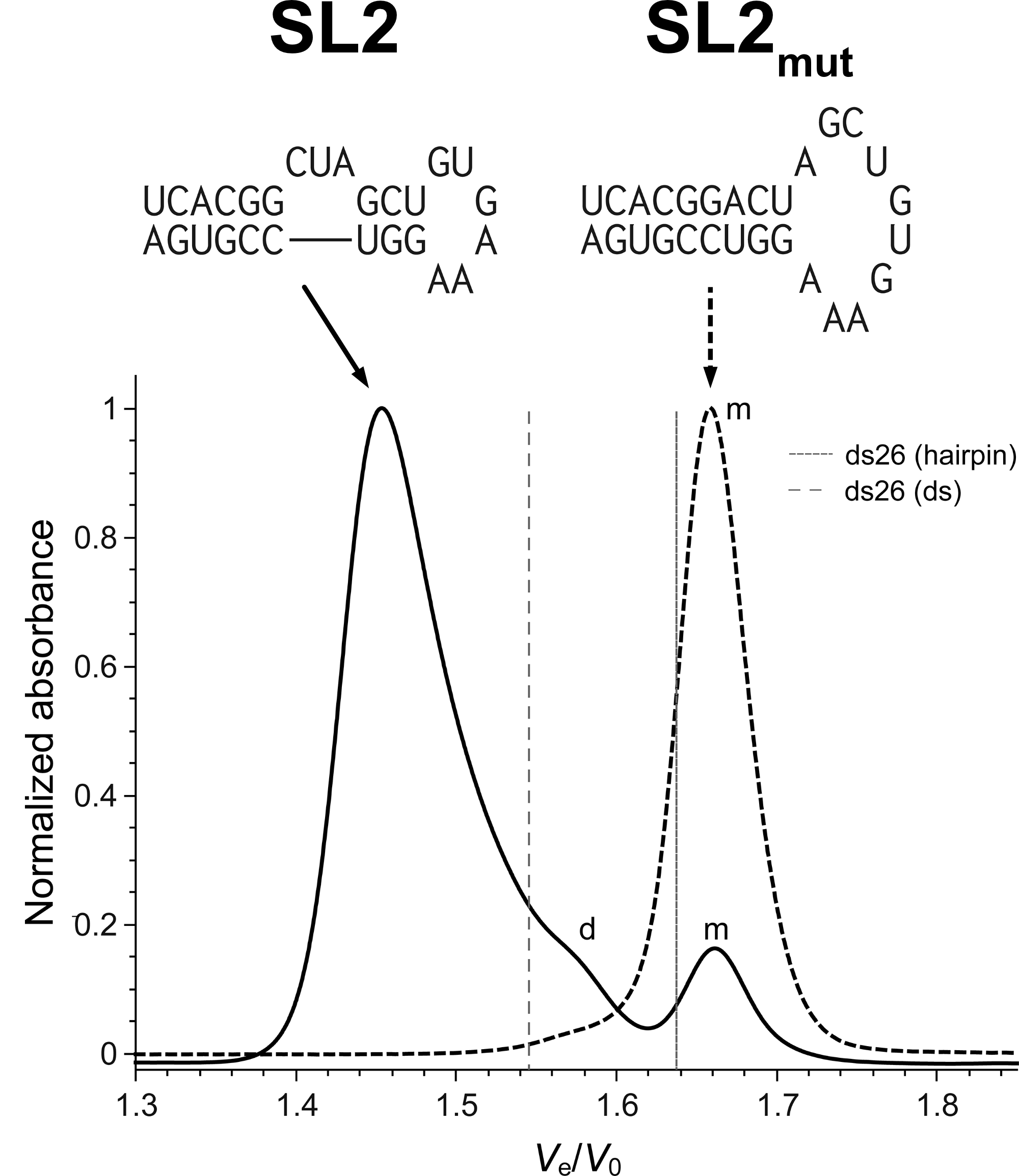

SEC allows robust stoichiometry determination
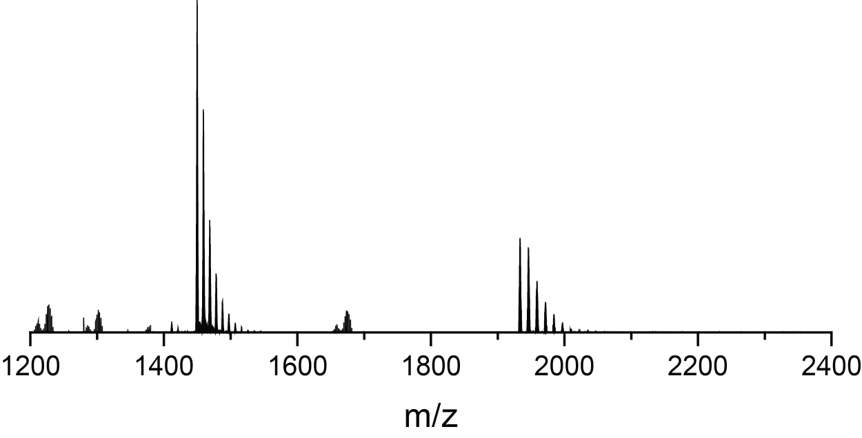
Native MS probes non-covalent interactions
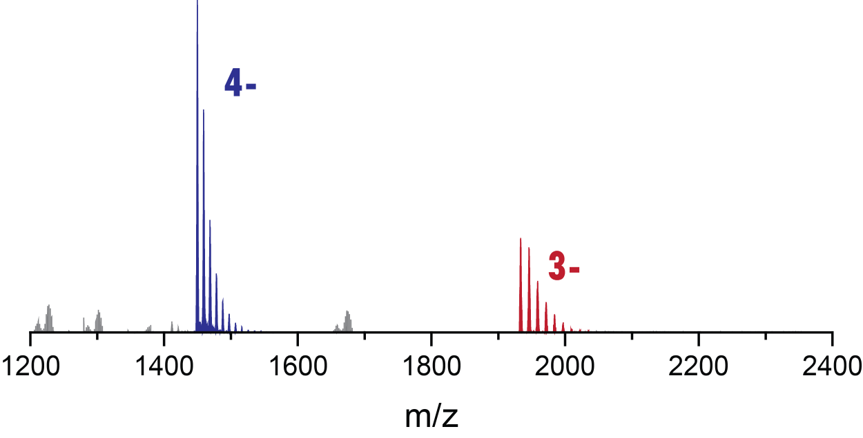
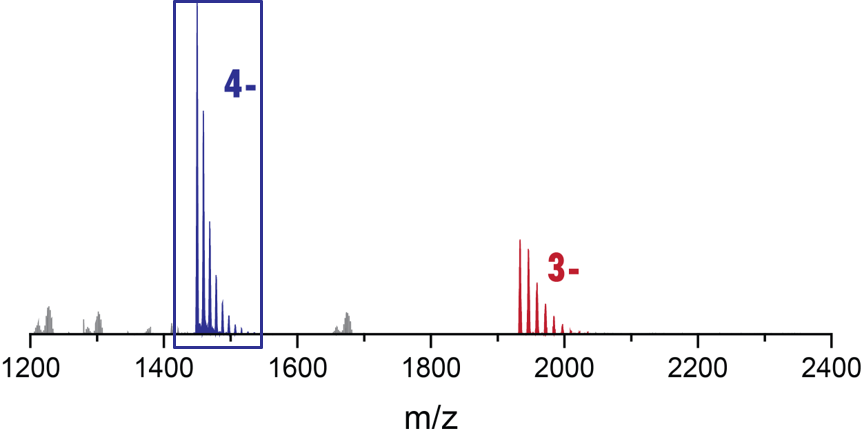
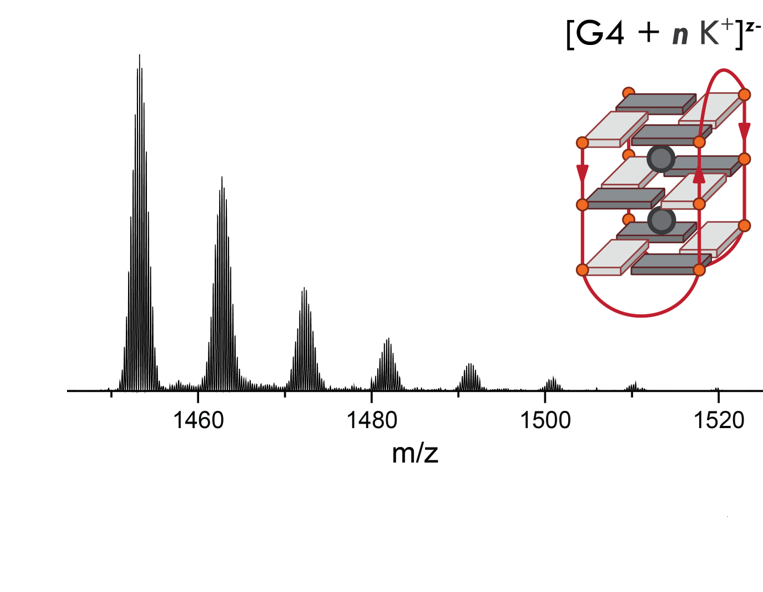
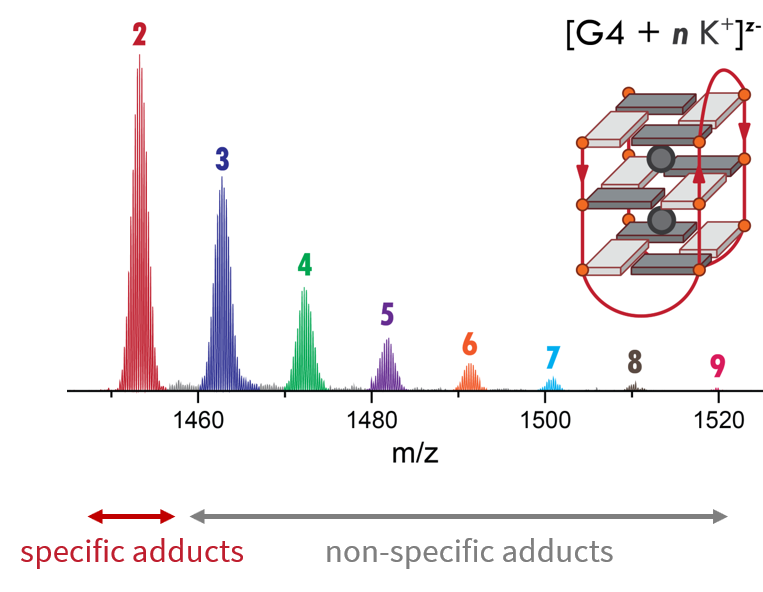
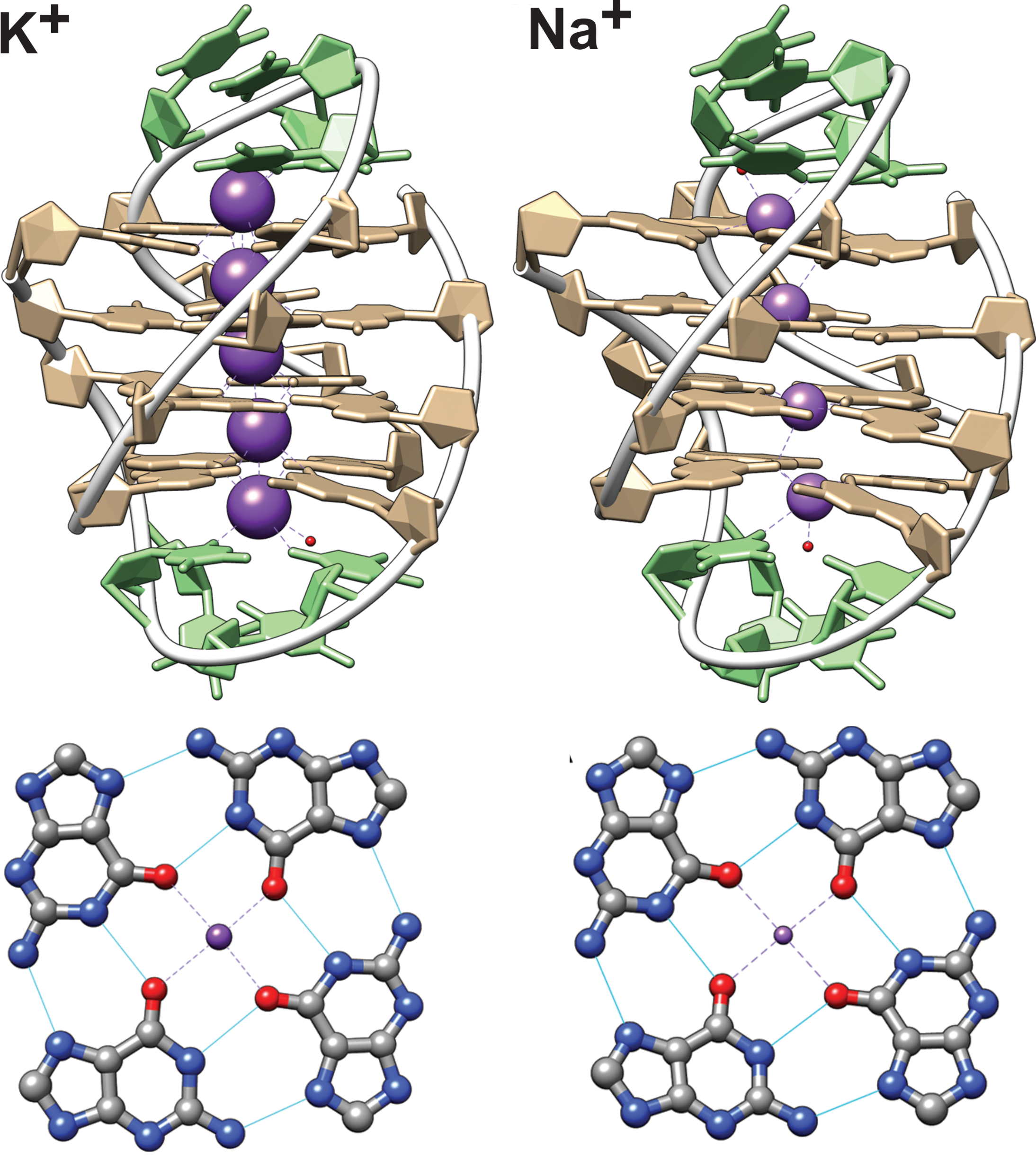
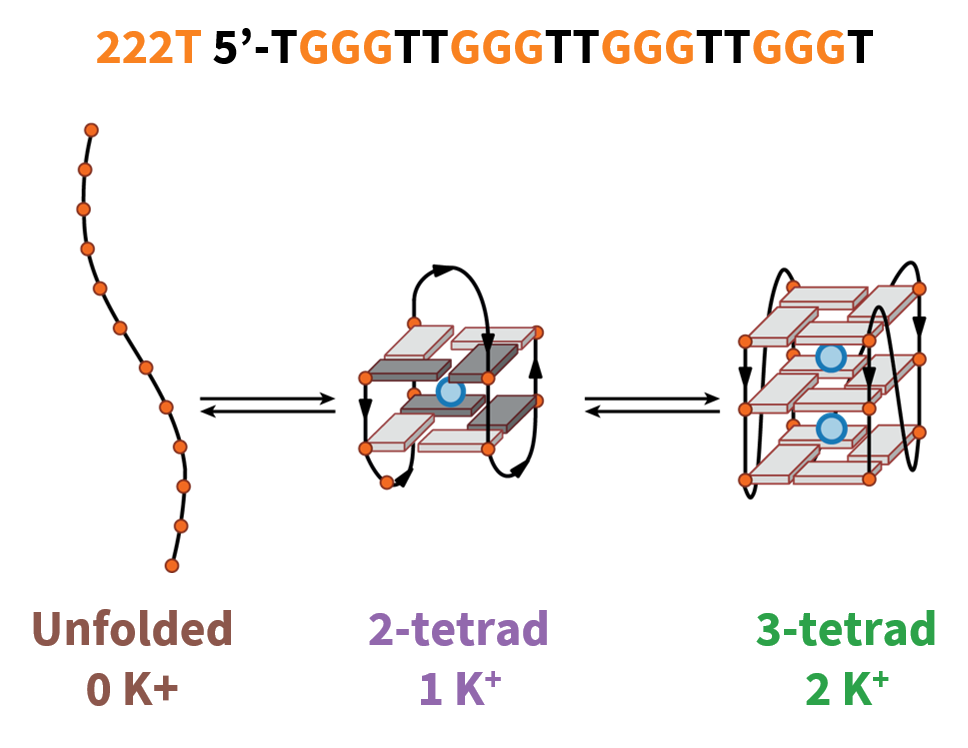
Adapting to a potassium-deficient buffer
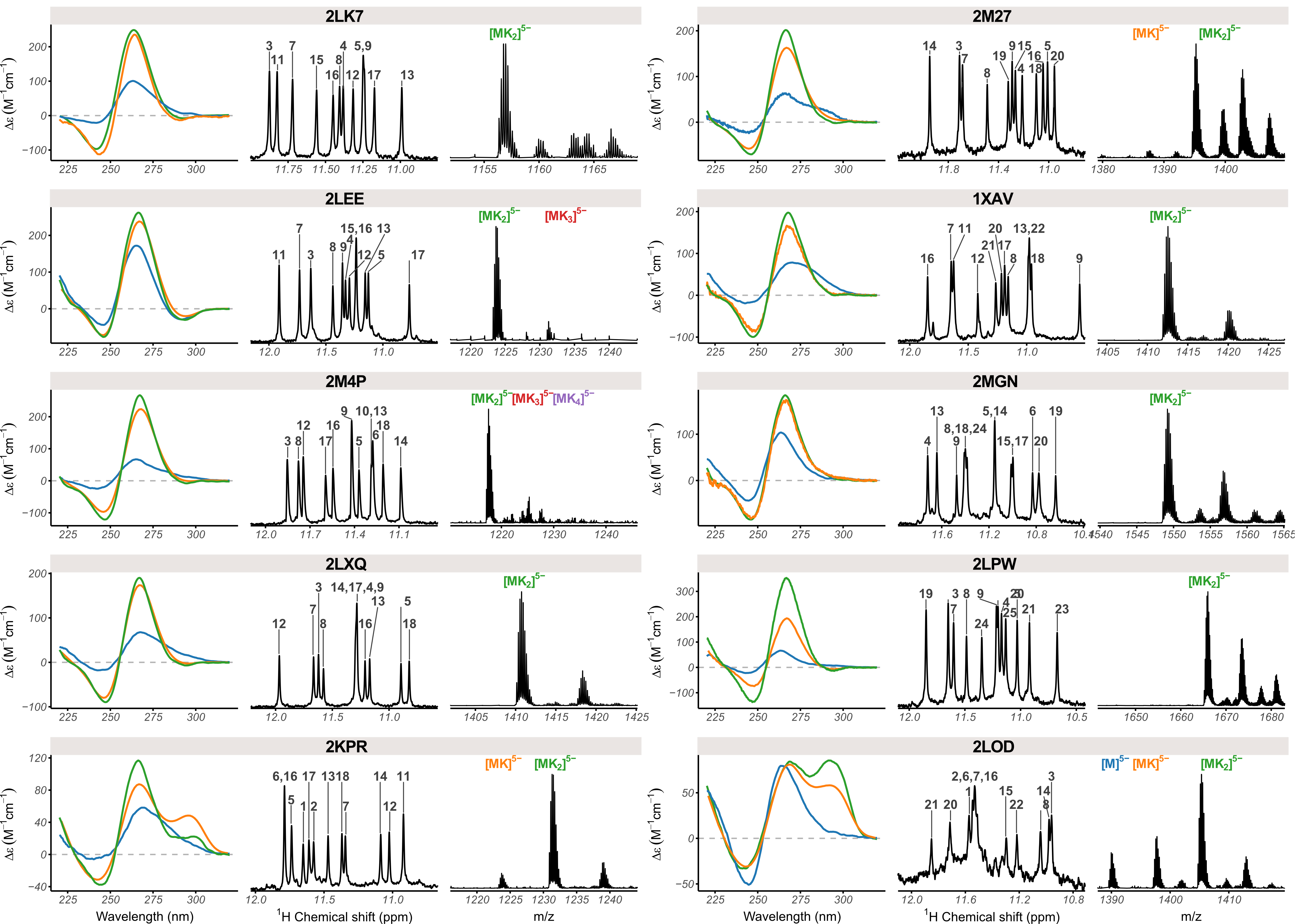

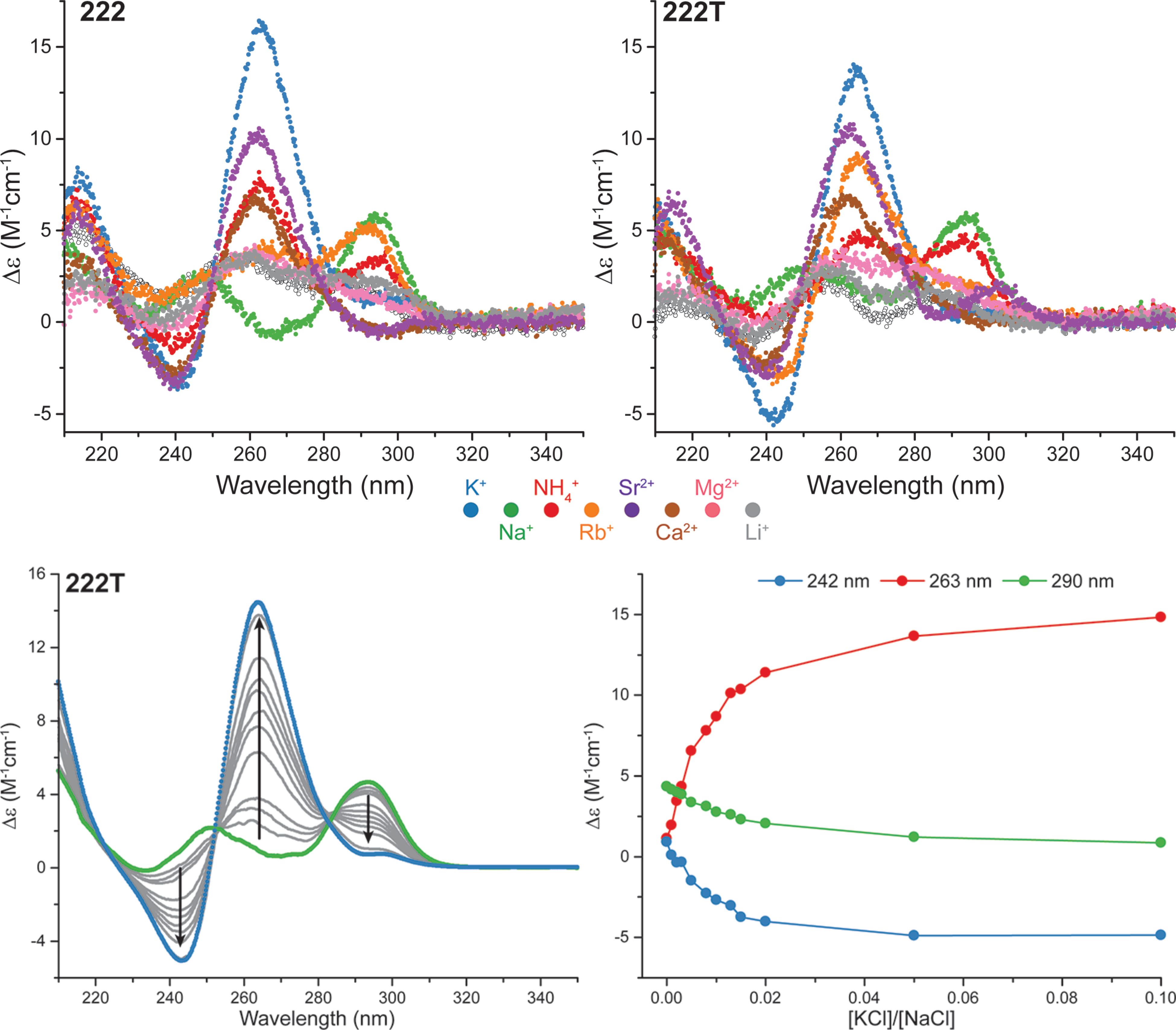
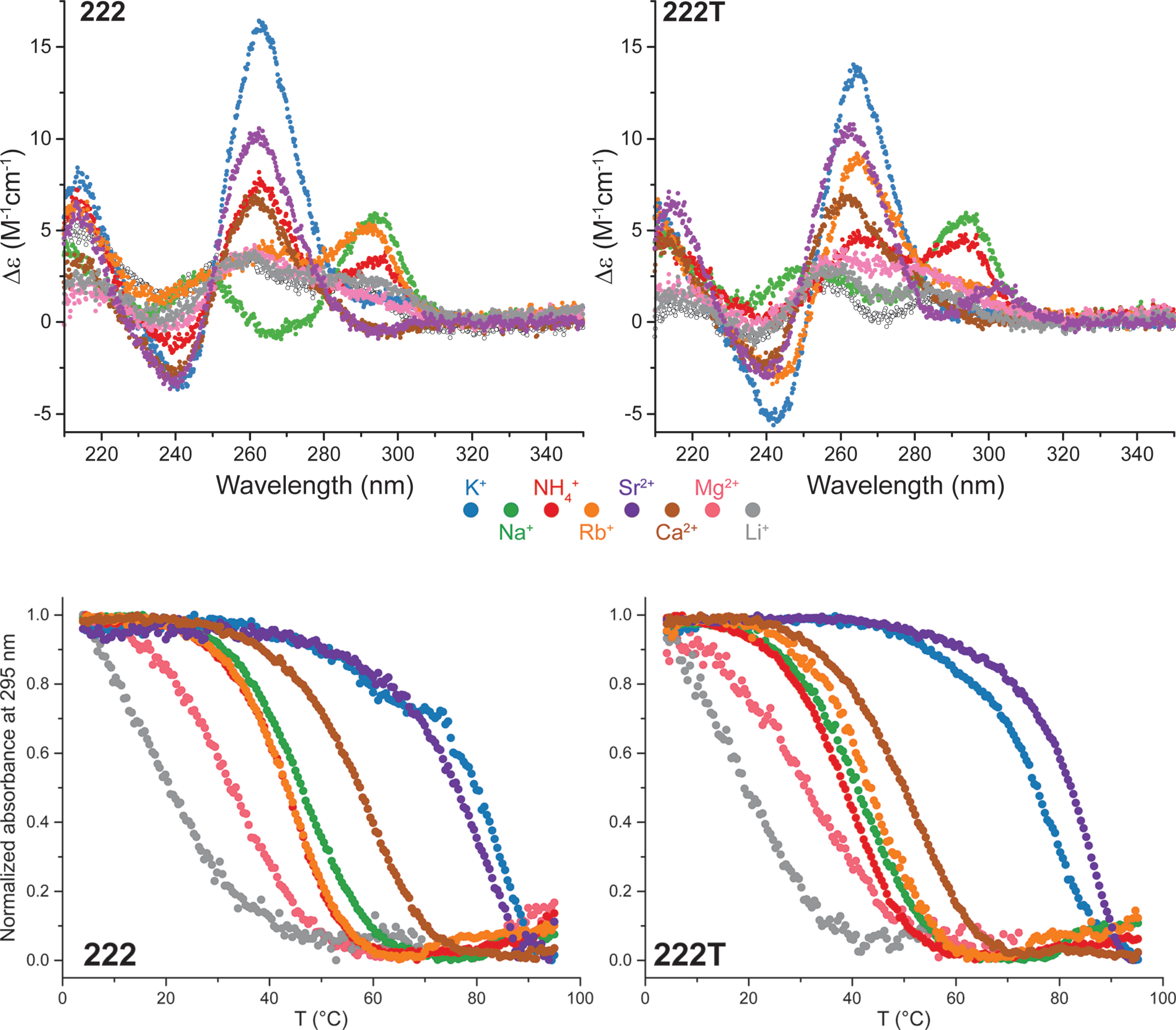
G4 conformations can be tuned with cations
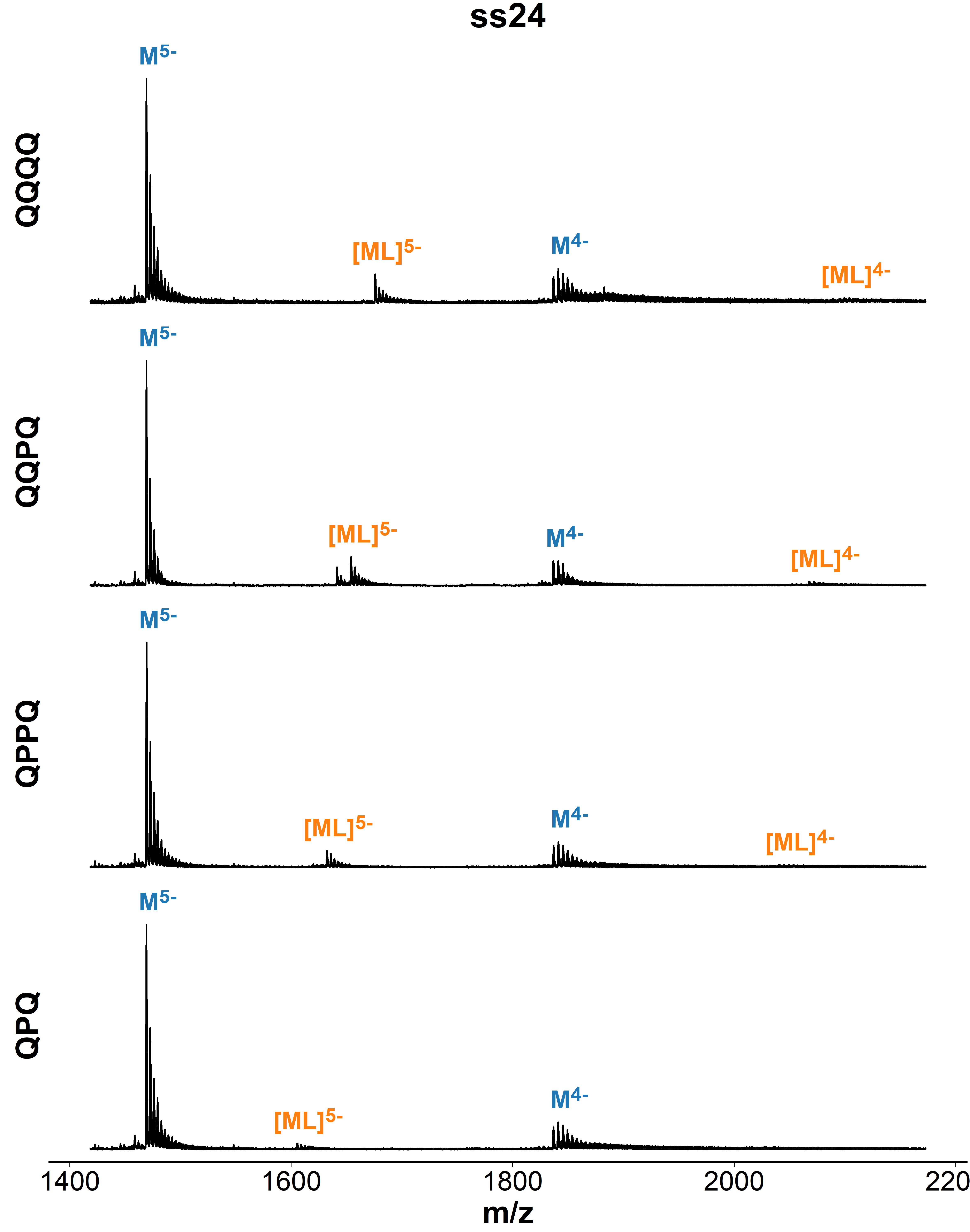
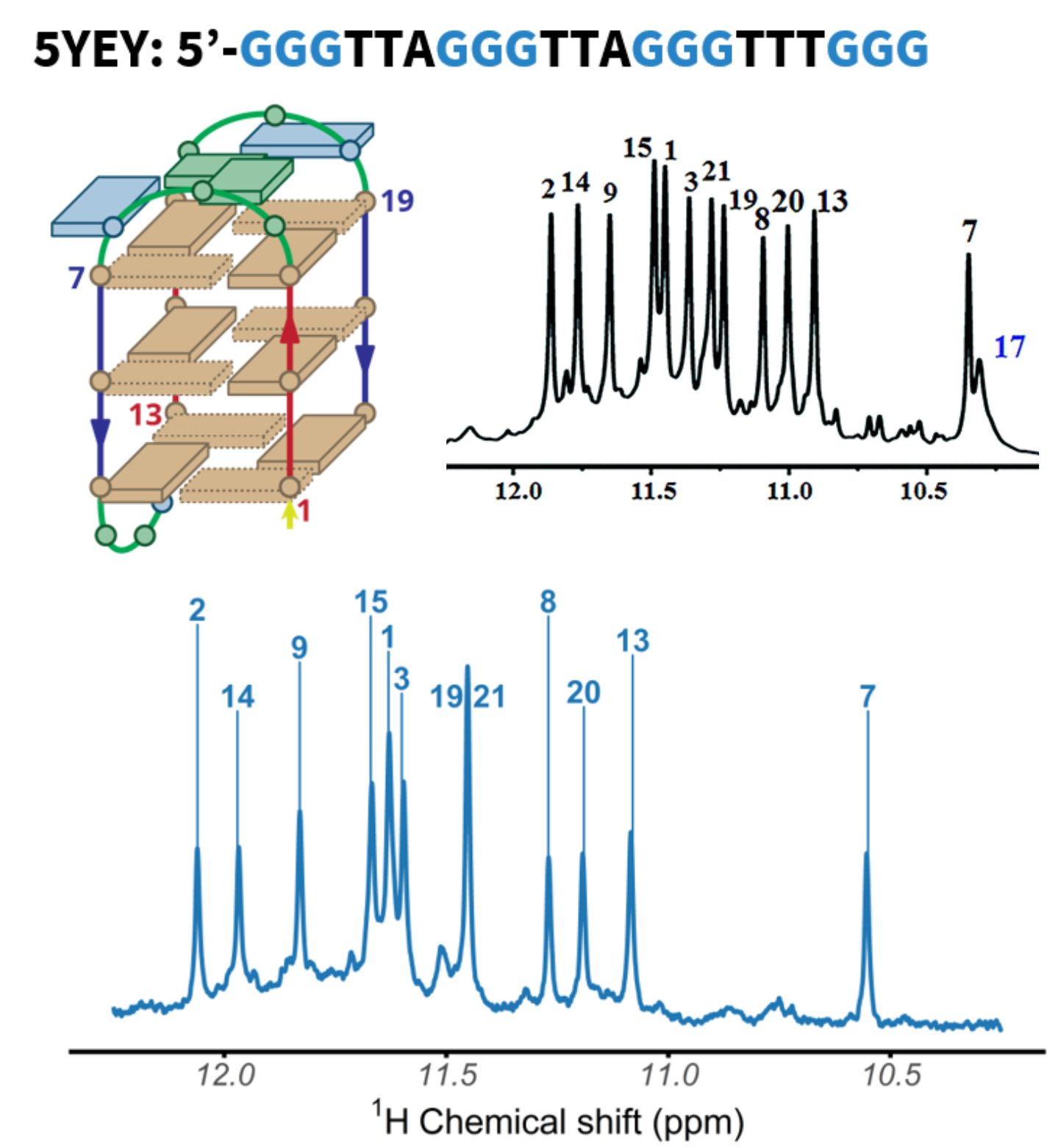
222T: TGGGTTGGGTTGGGTTGGGT
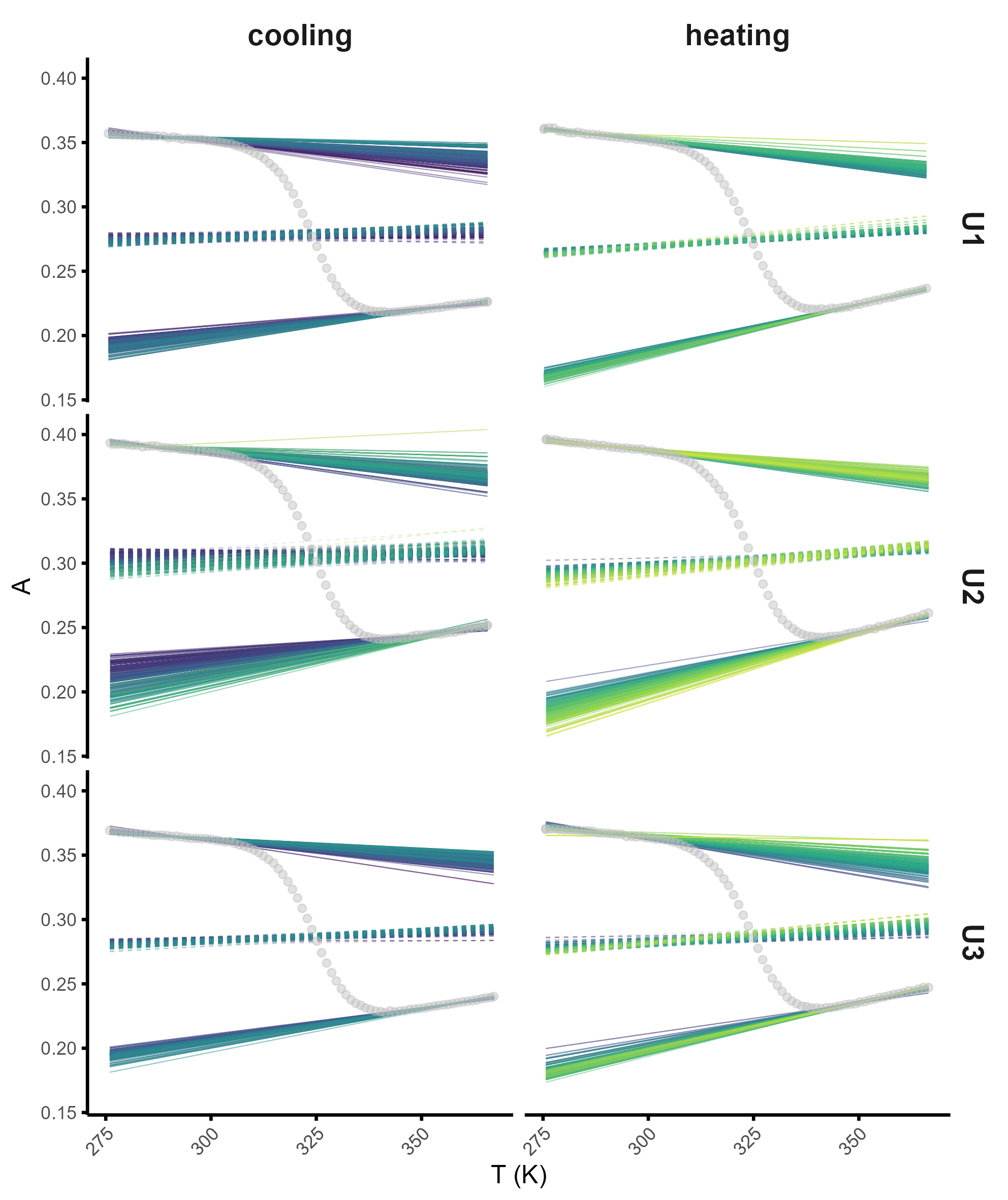
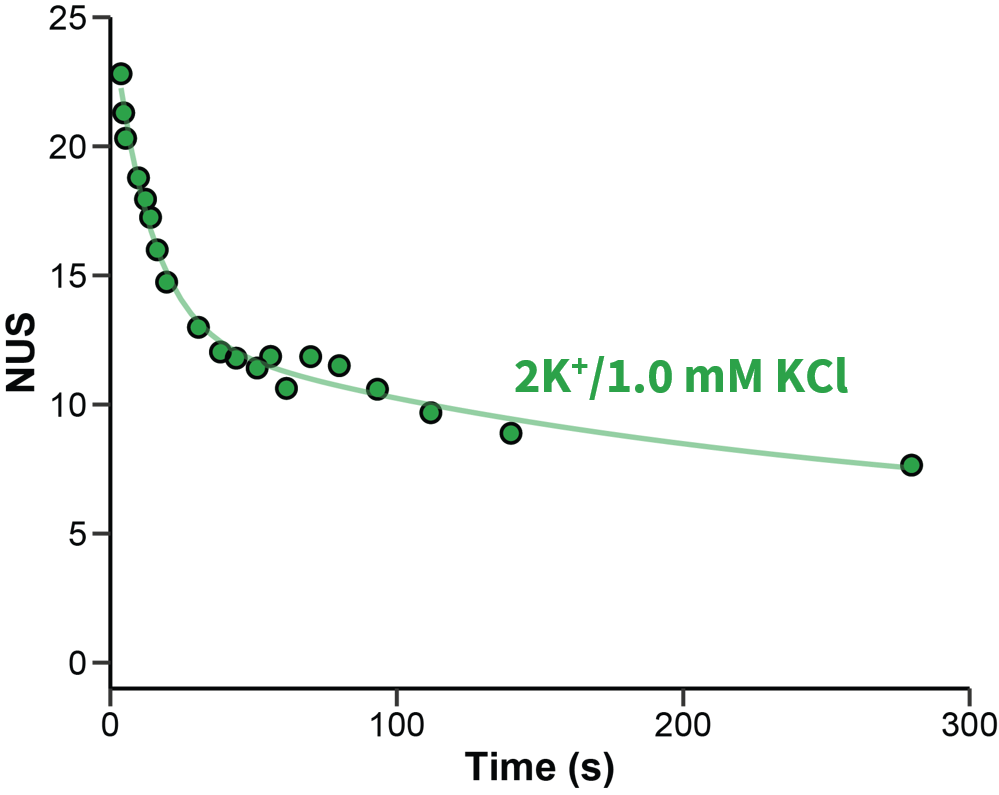
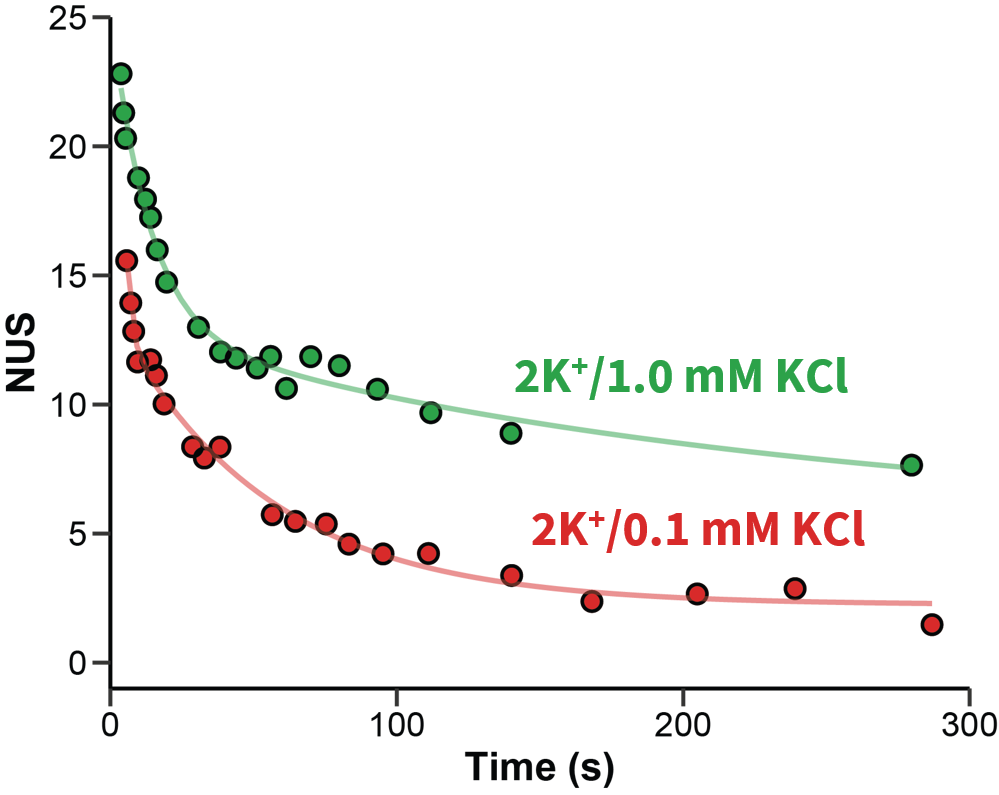
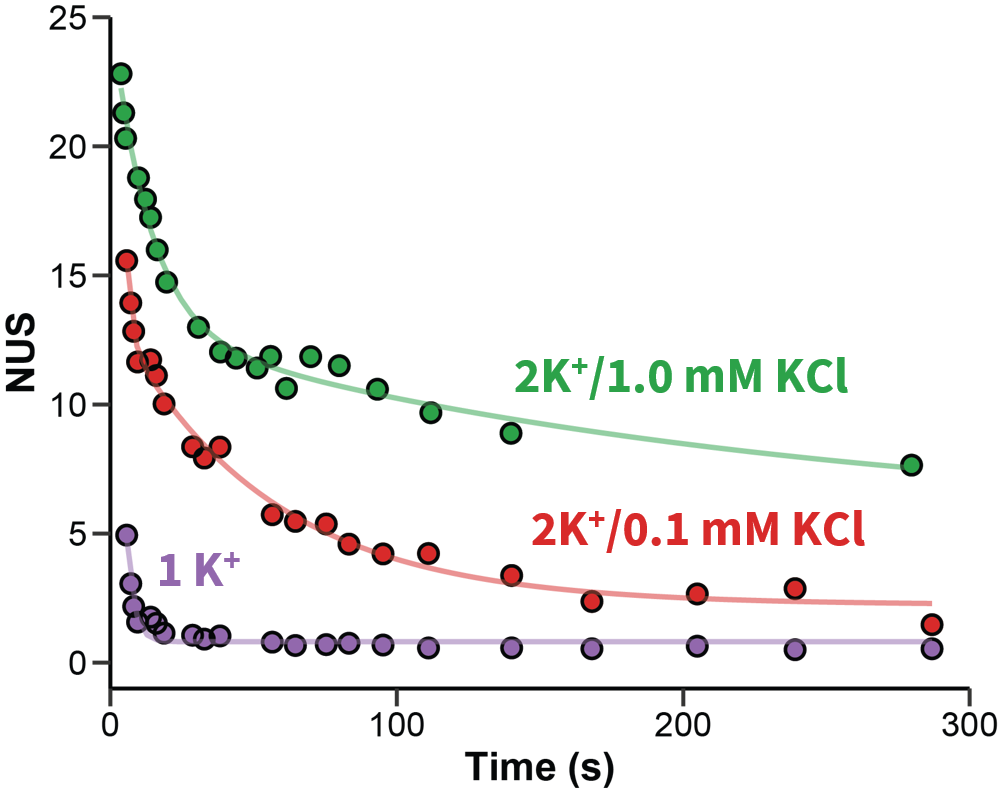
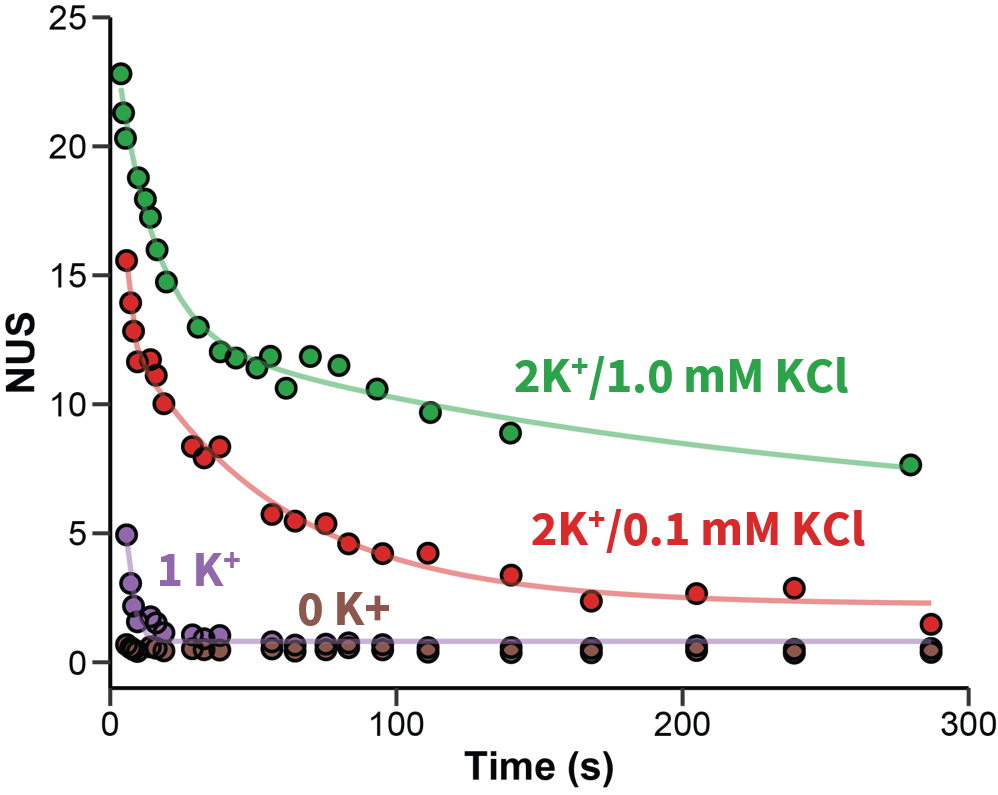
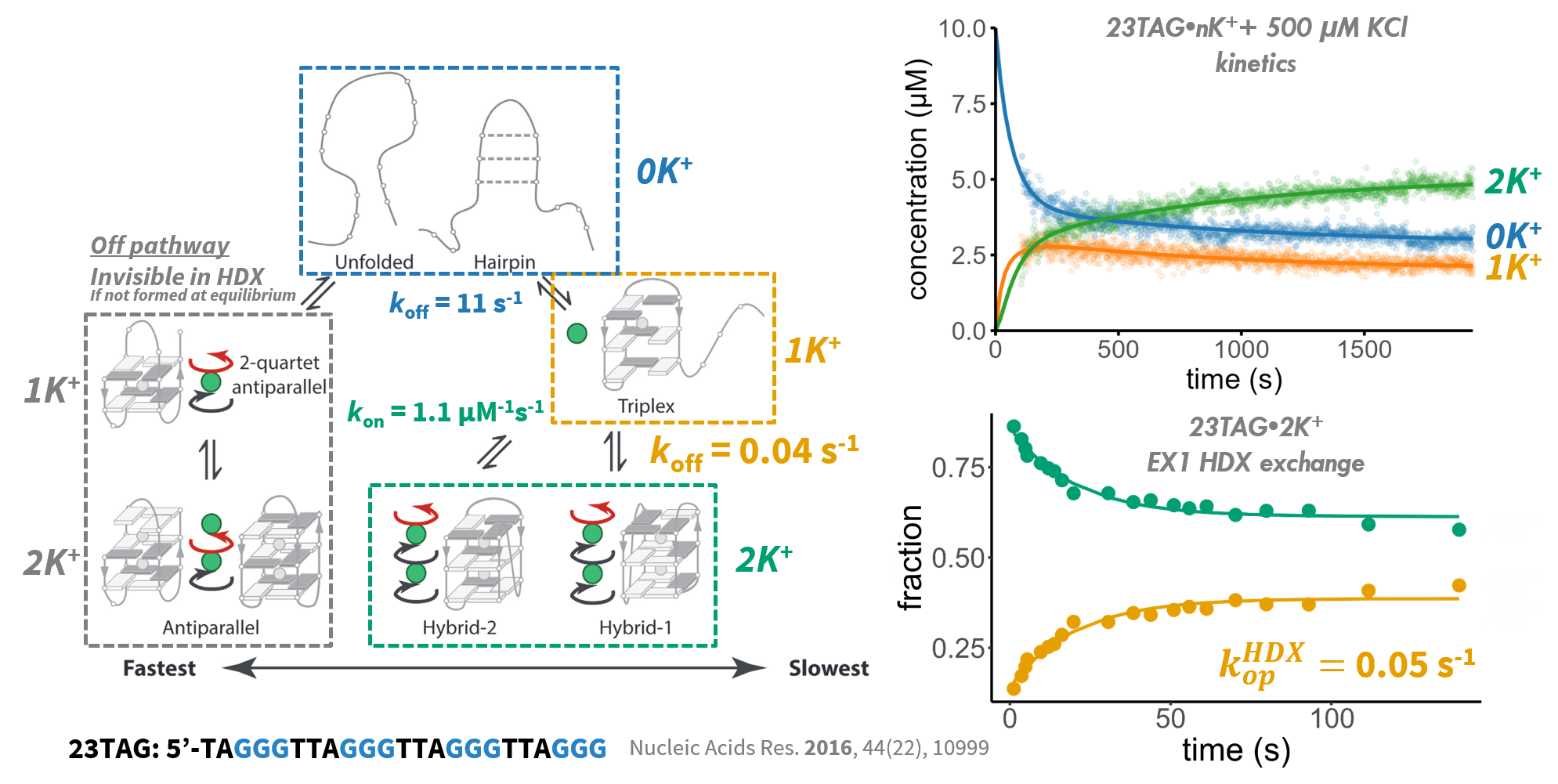
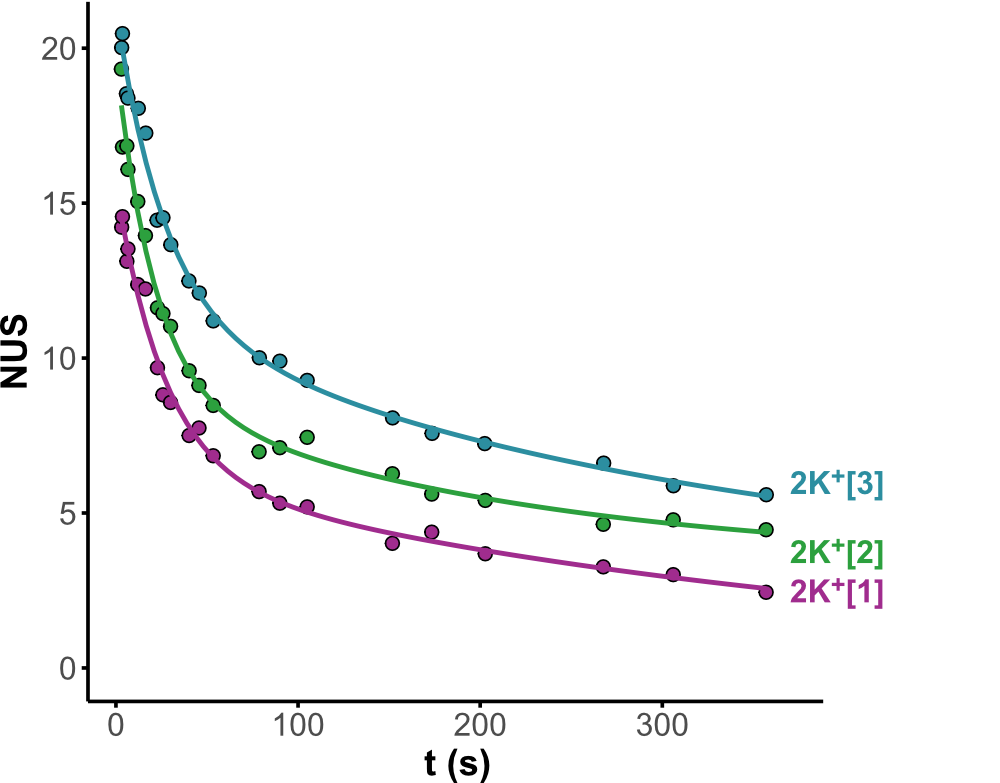
The interconversion occurs at low [KCl]
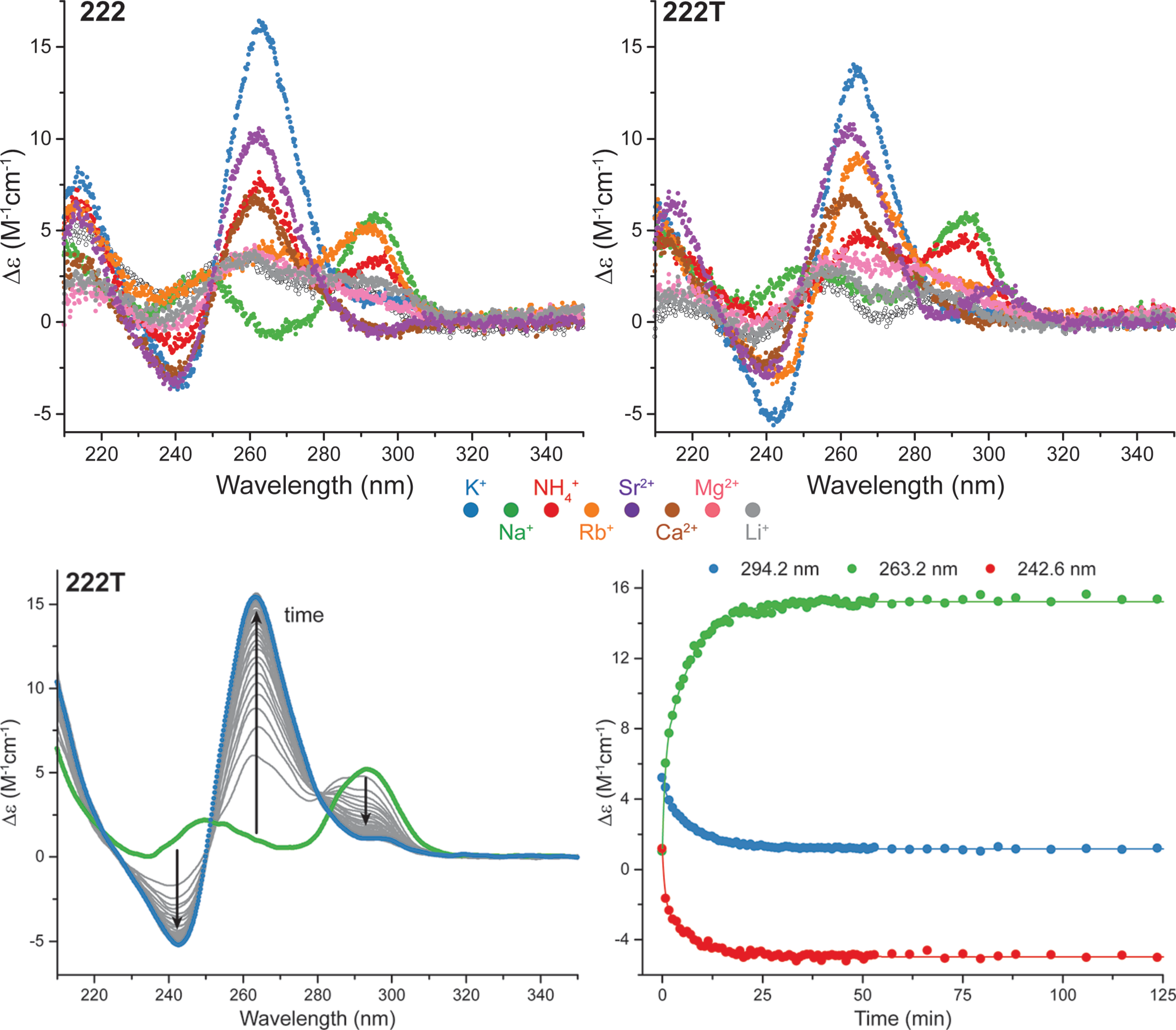
The interconversion is fast
The interconversion arises from large ΔTm
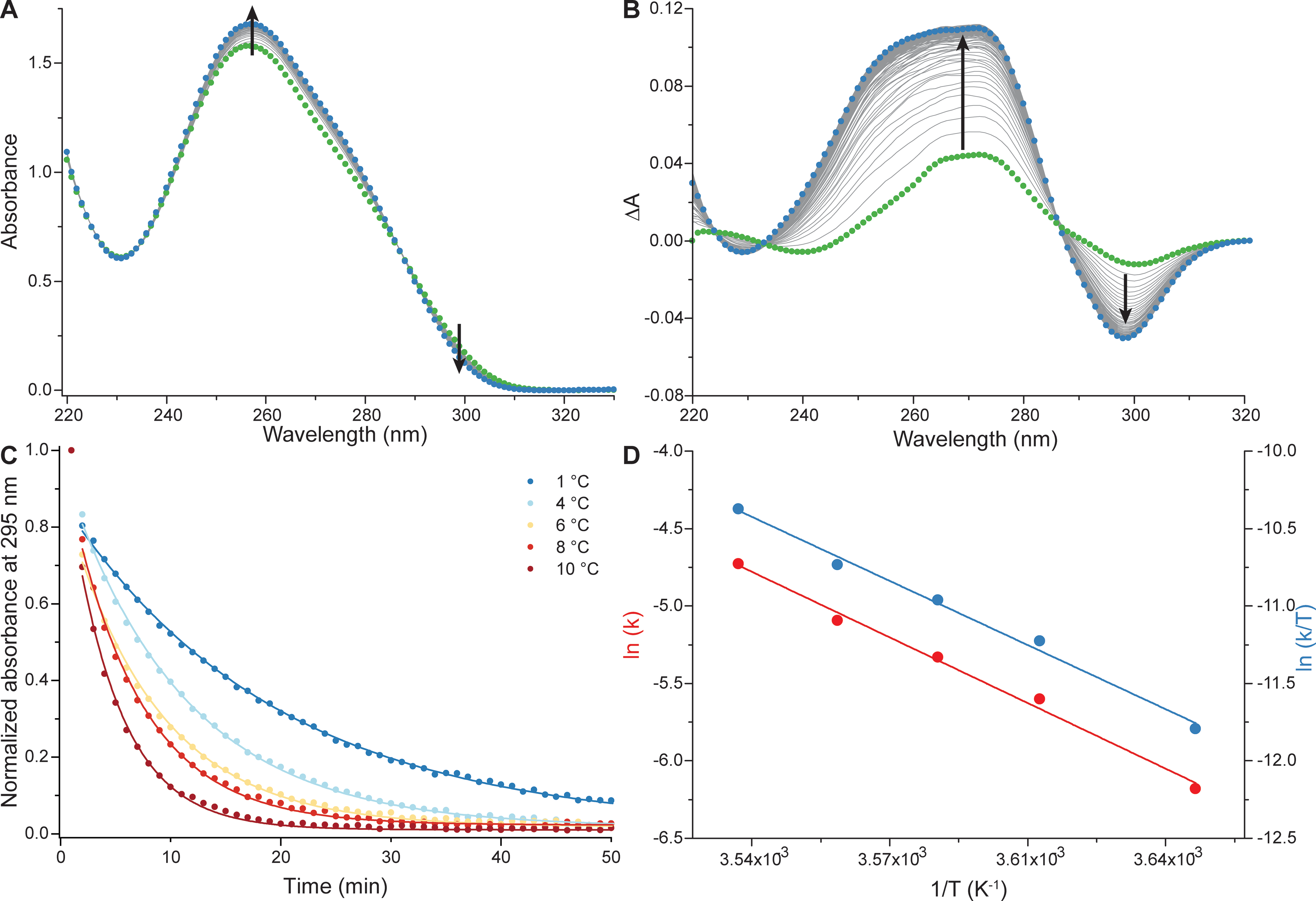
The interconversion can be monitored by UV
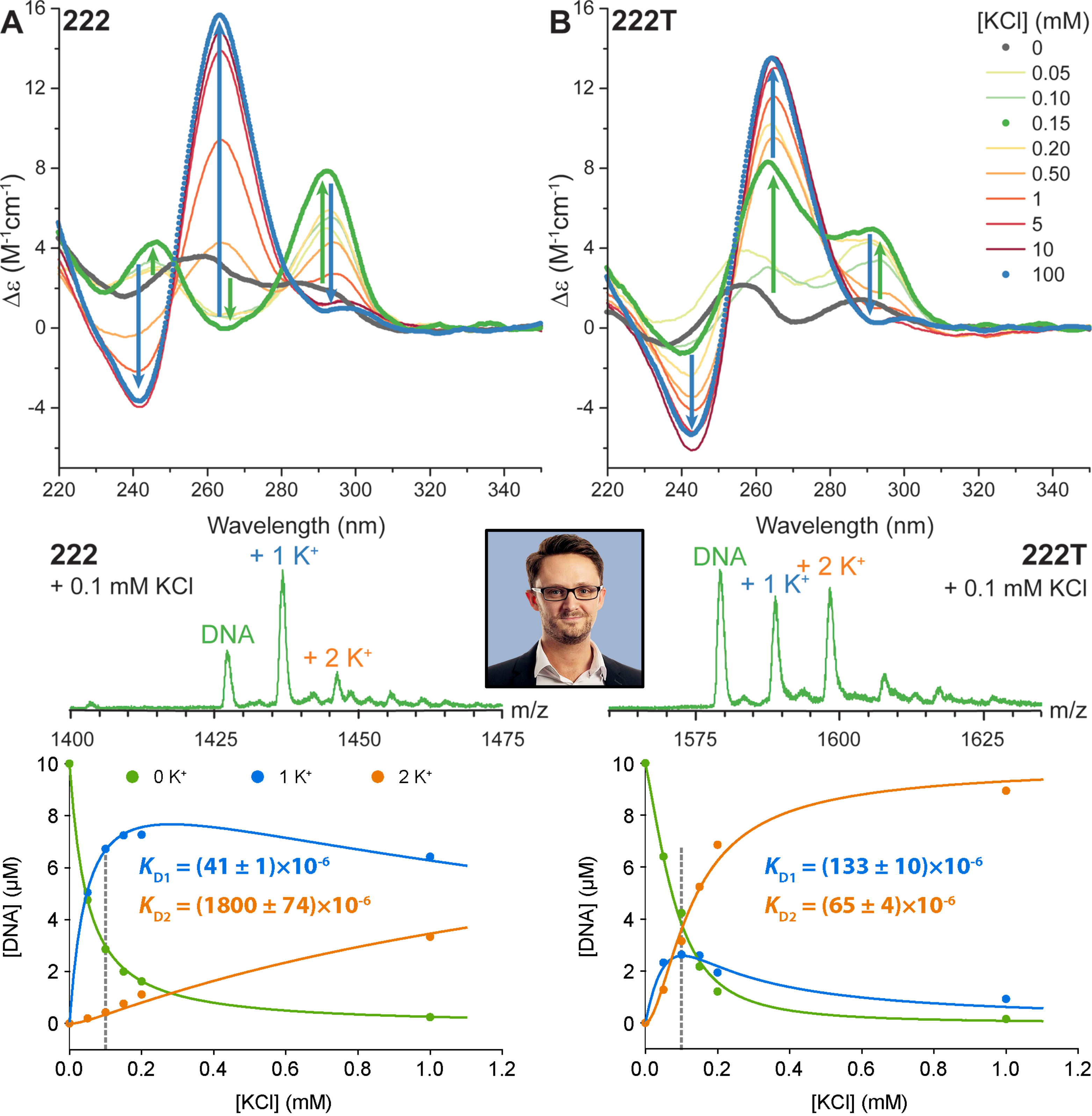
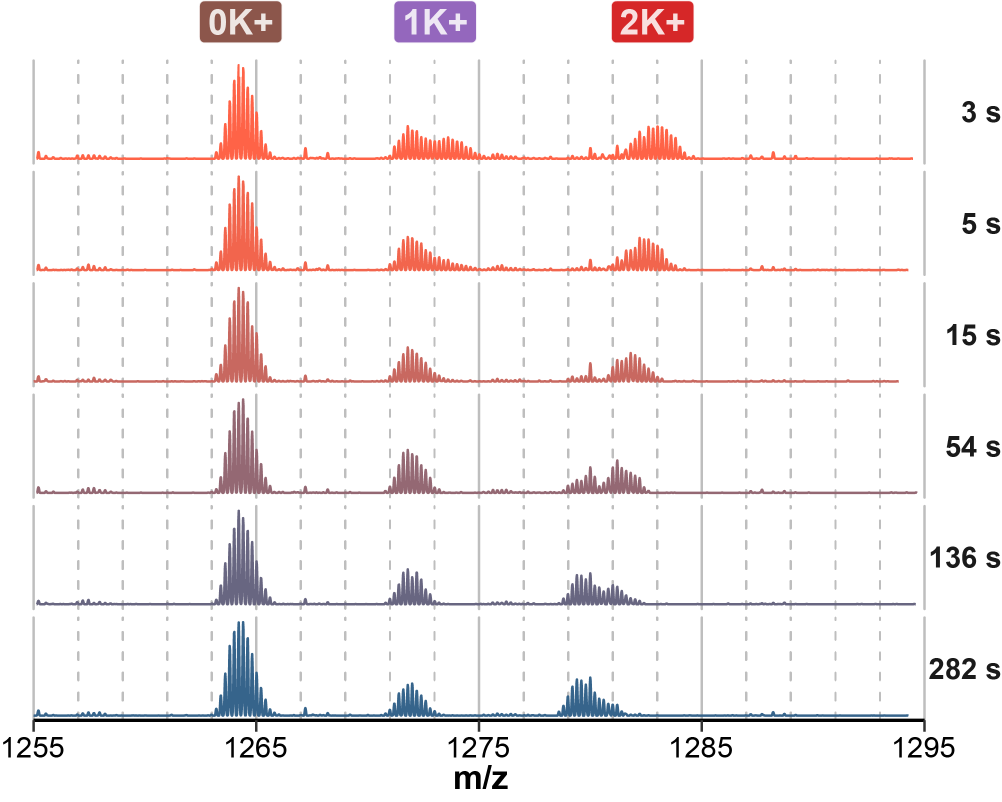
The interconversion can be monitored by nMS

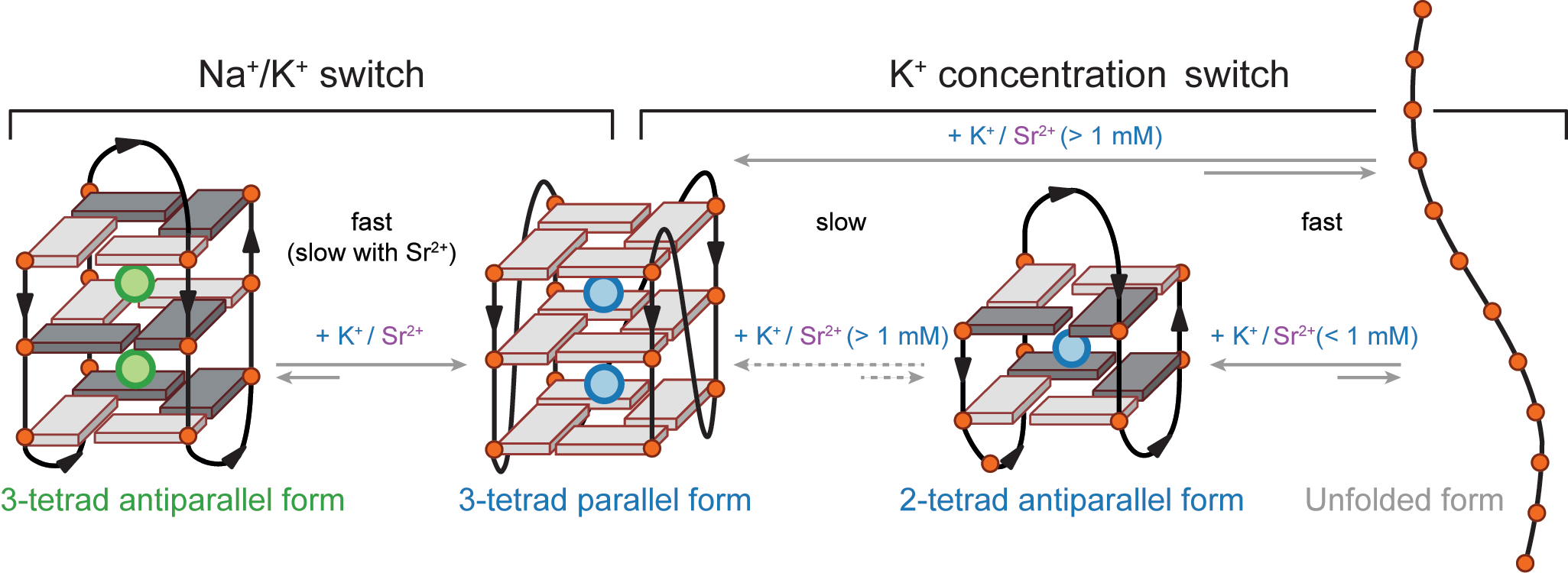
The combination of methods allows thermodynamic
and kinetic characterization of complex system
G4 can be targeted by small molecules

Most G4 ligands are \(\pi\)-stacking on external tetrads
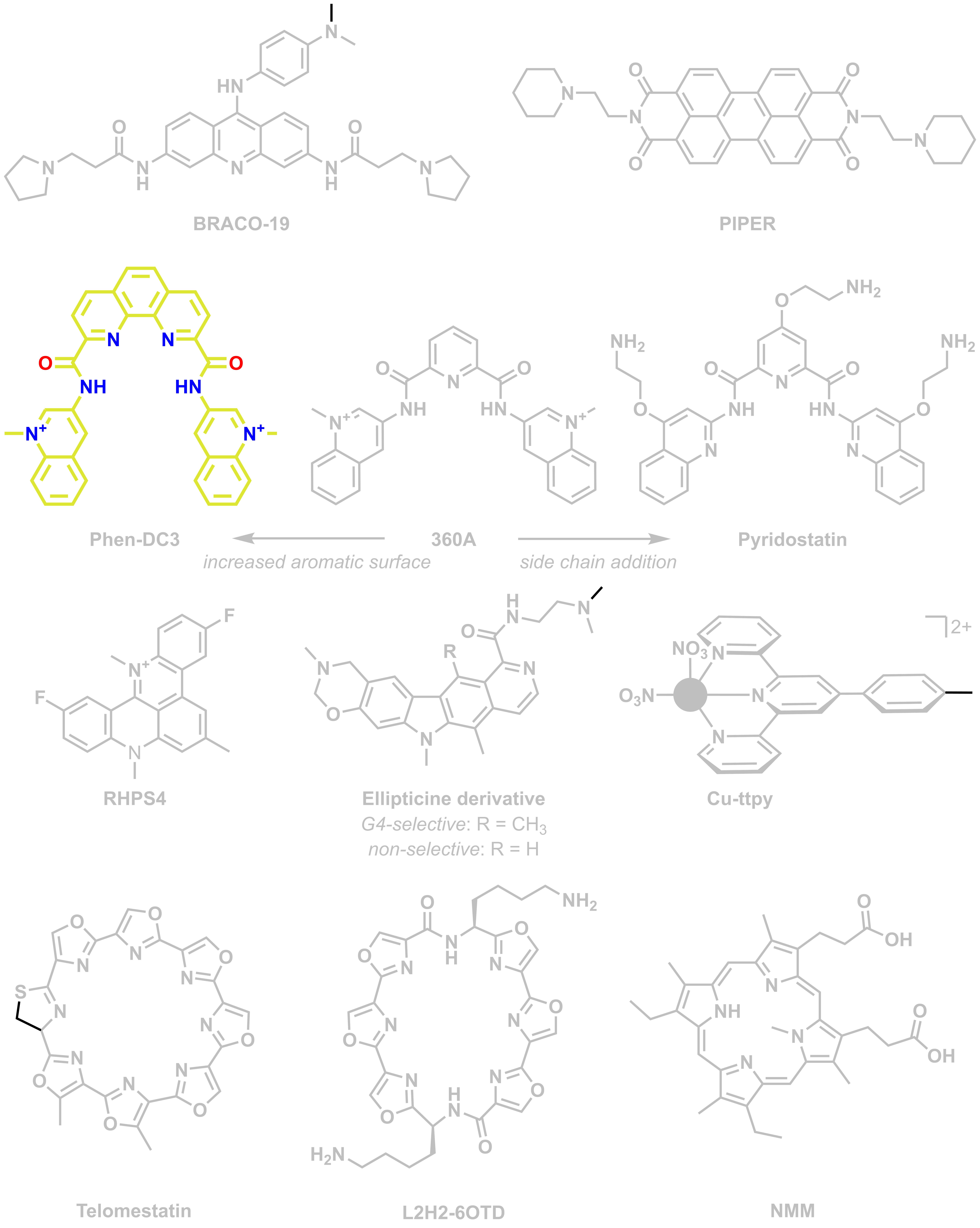
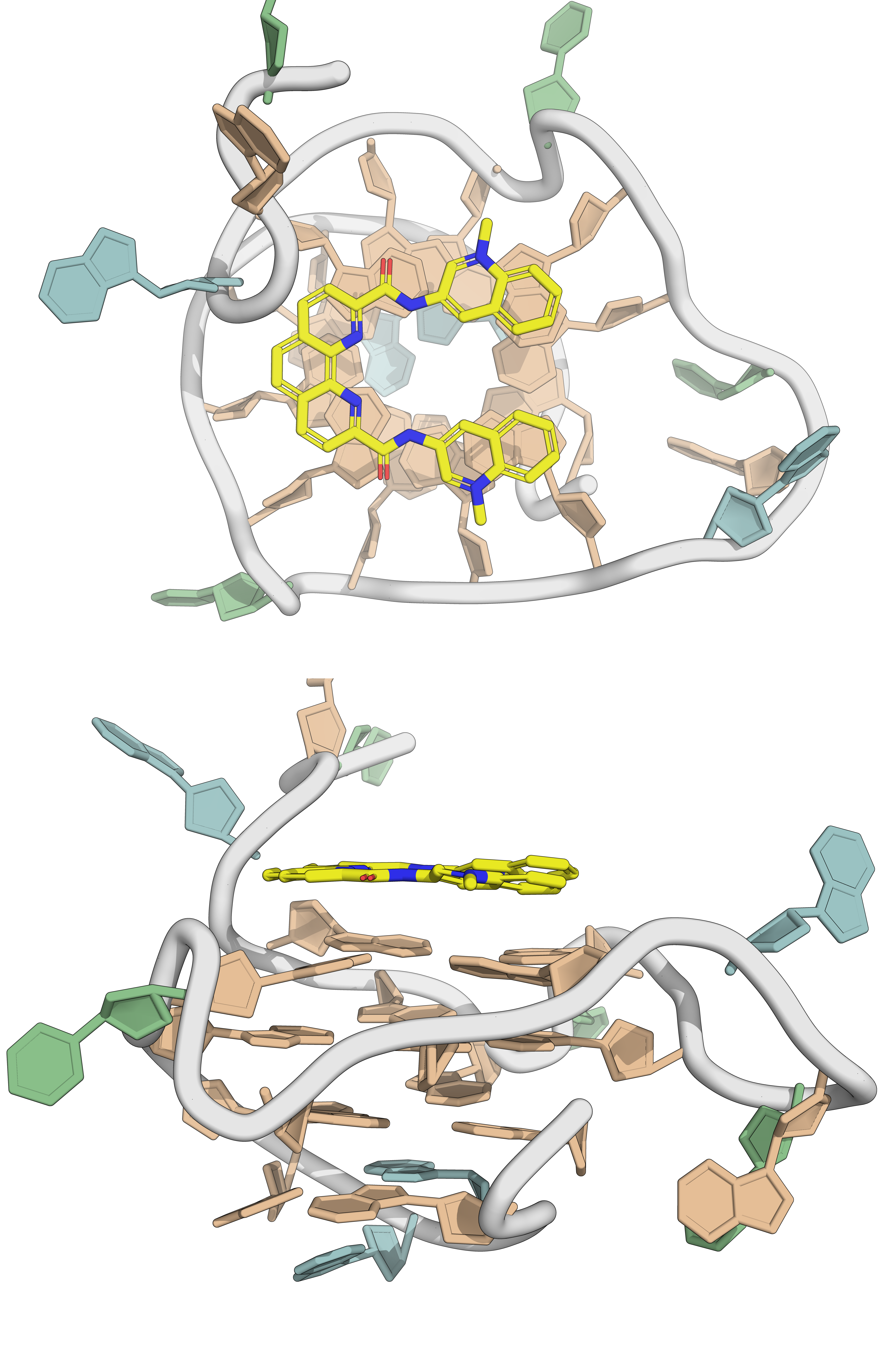
Sometimes luck does better than reason

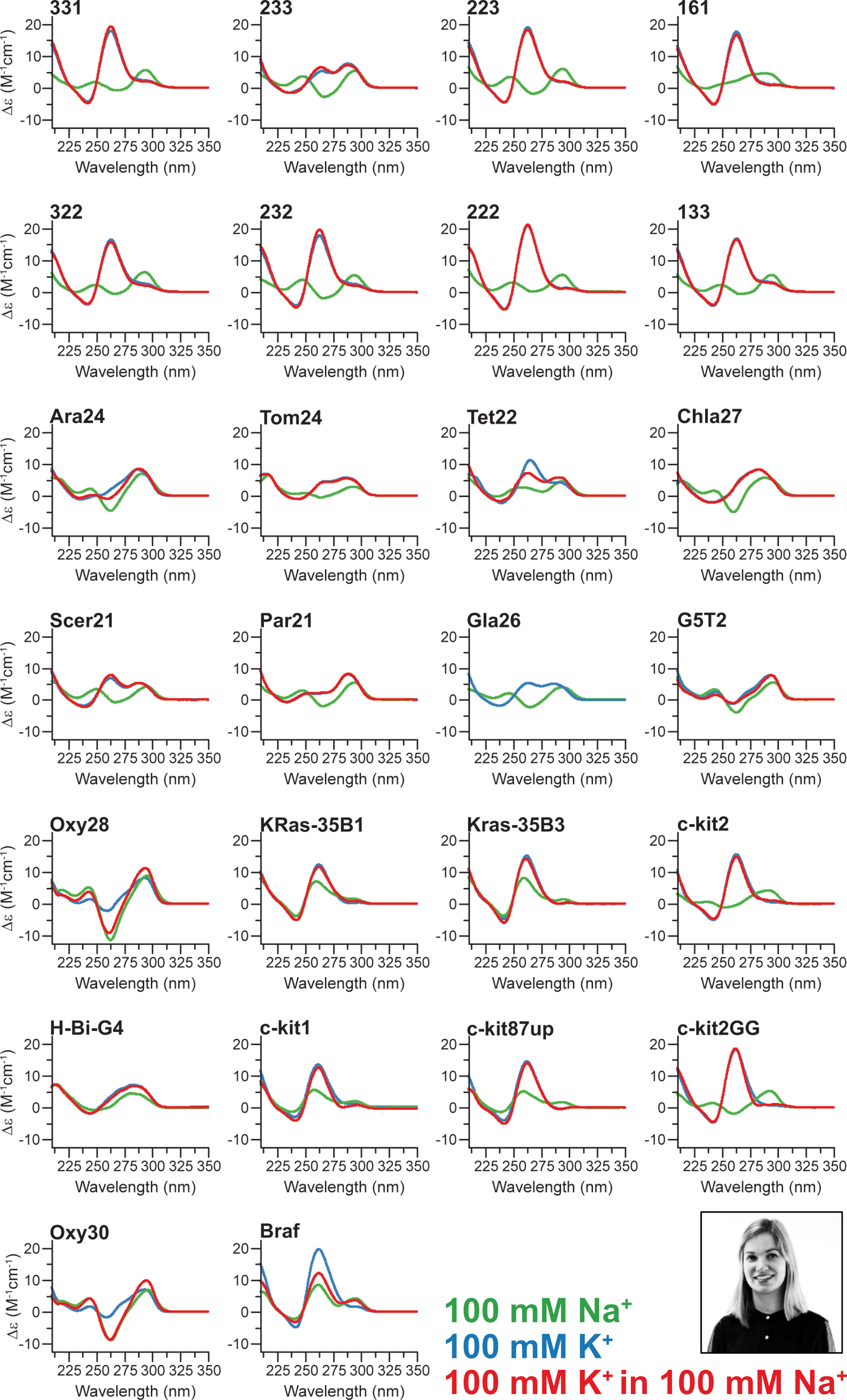
Binding preference to parallel topology
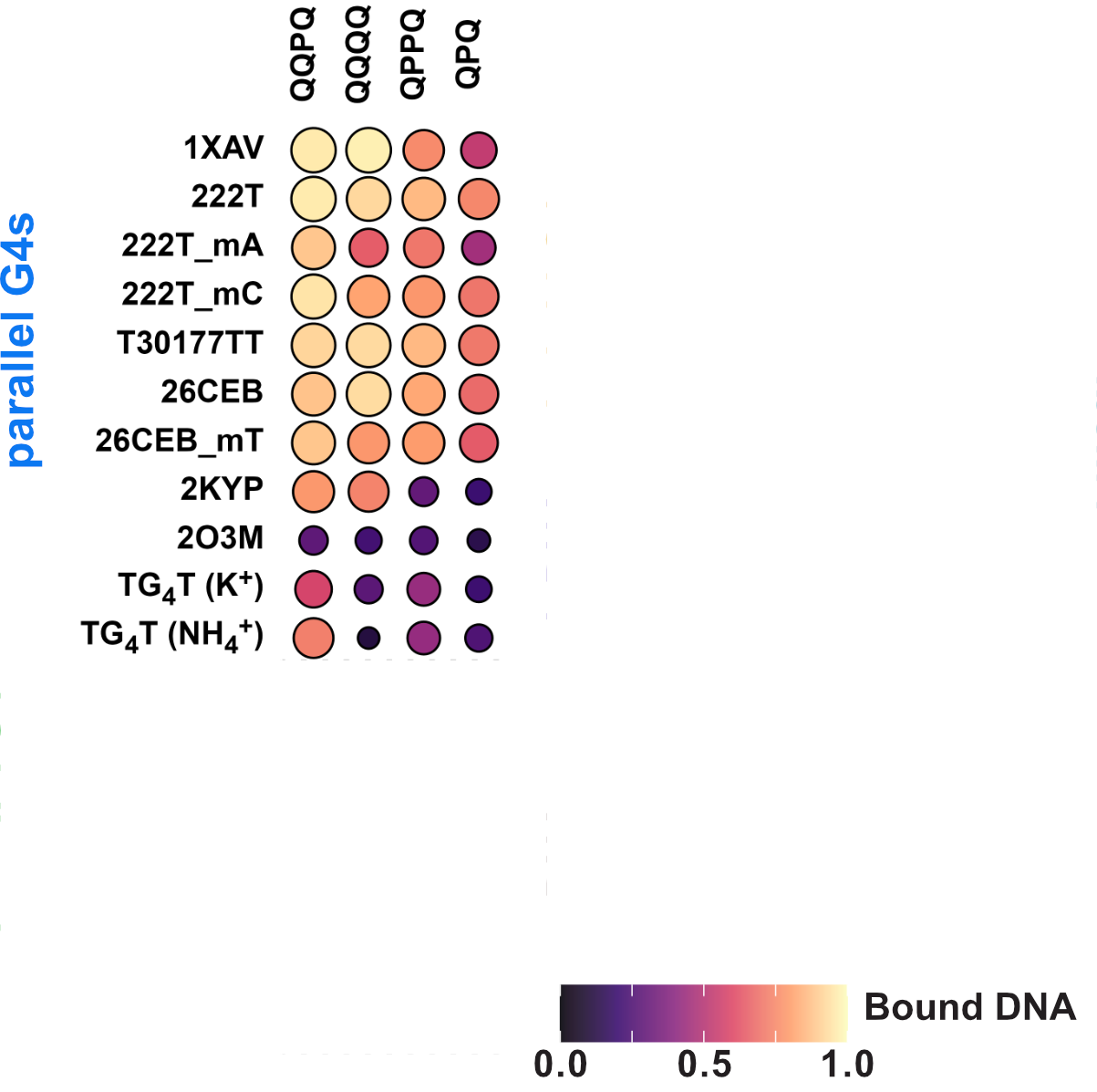
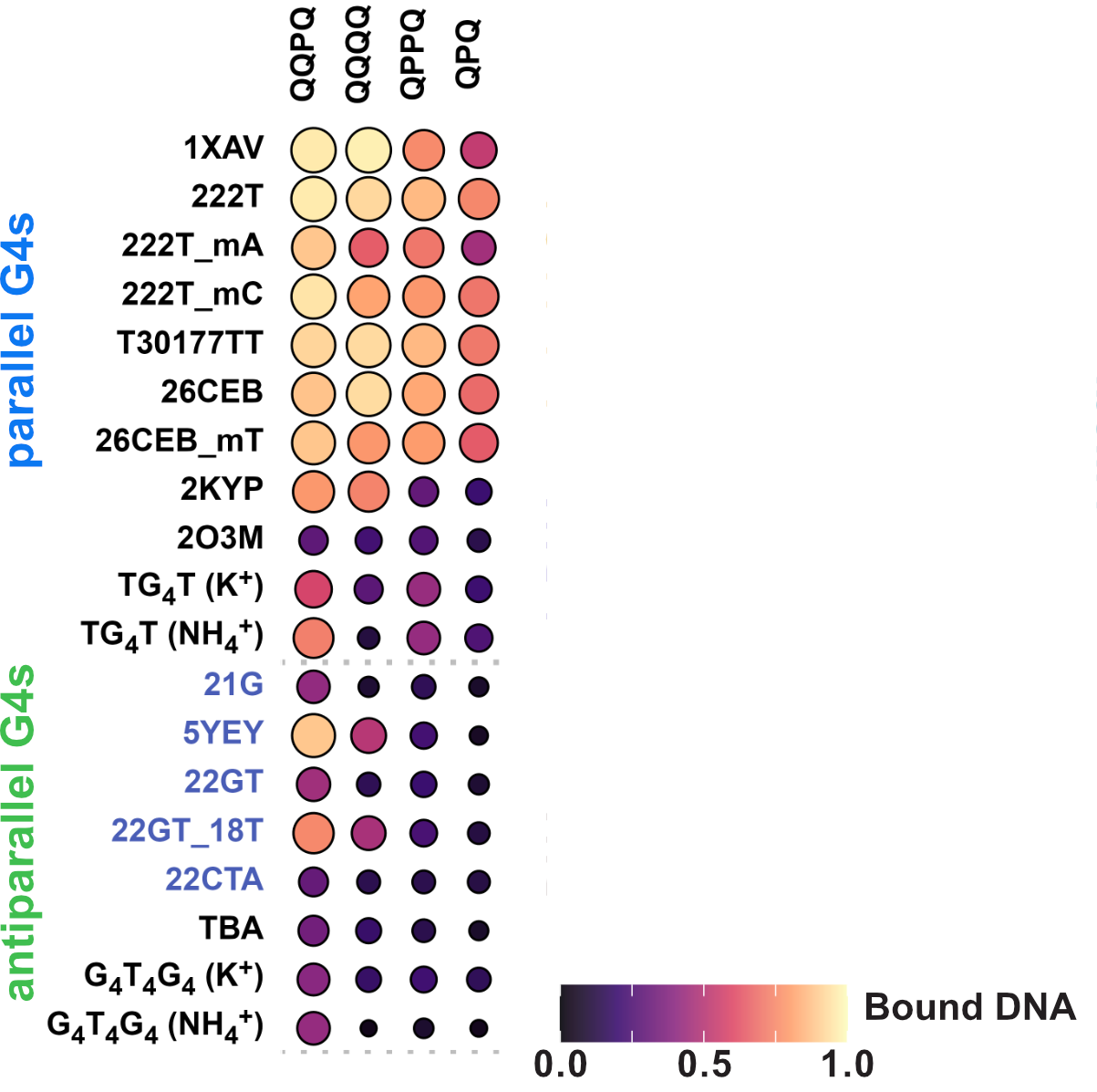
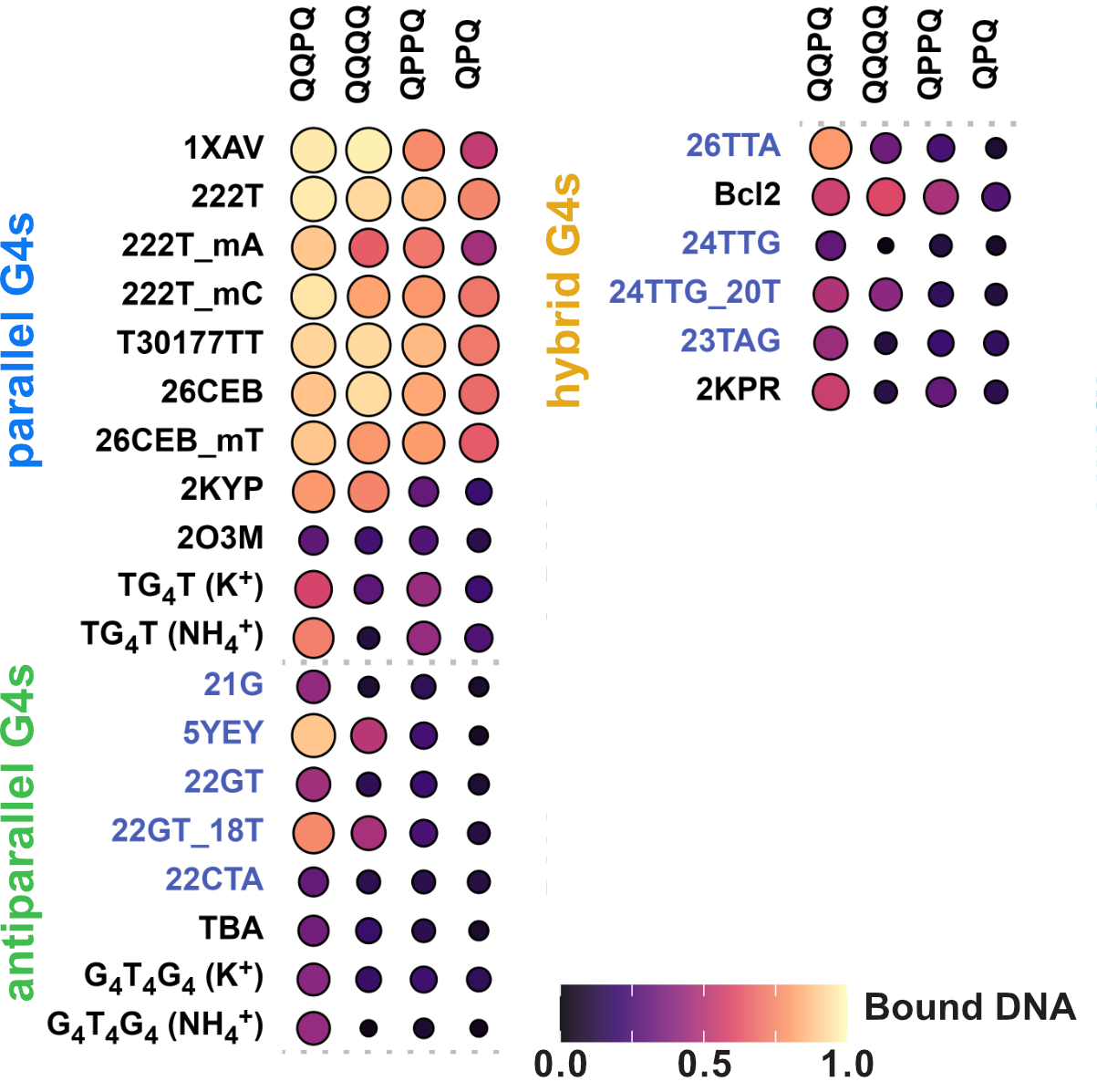
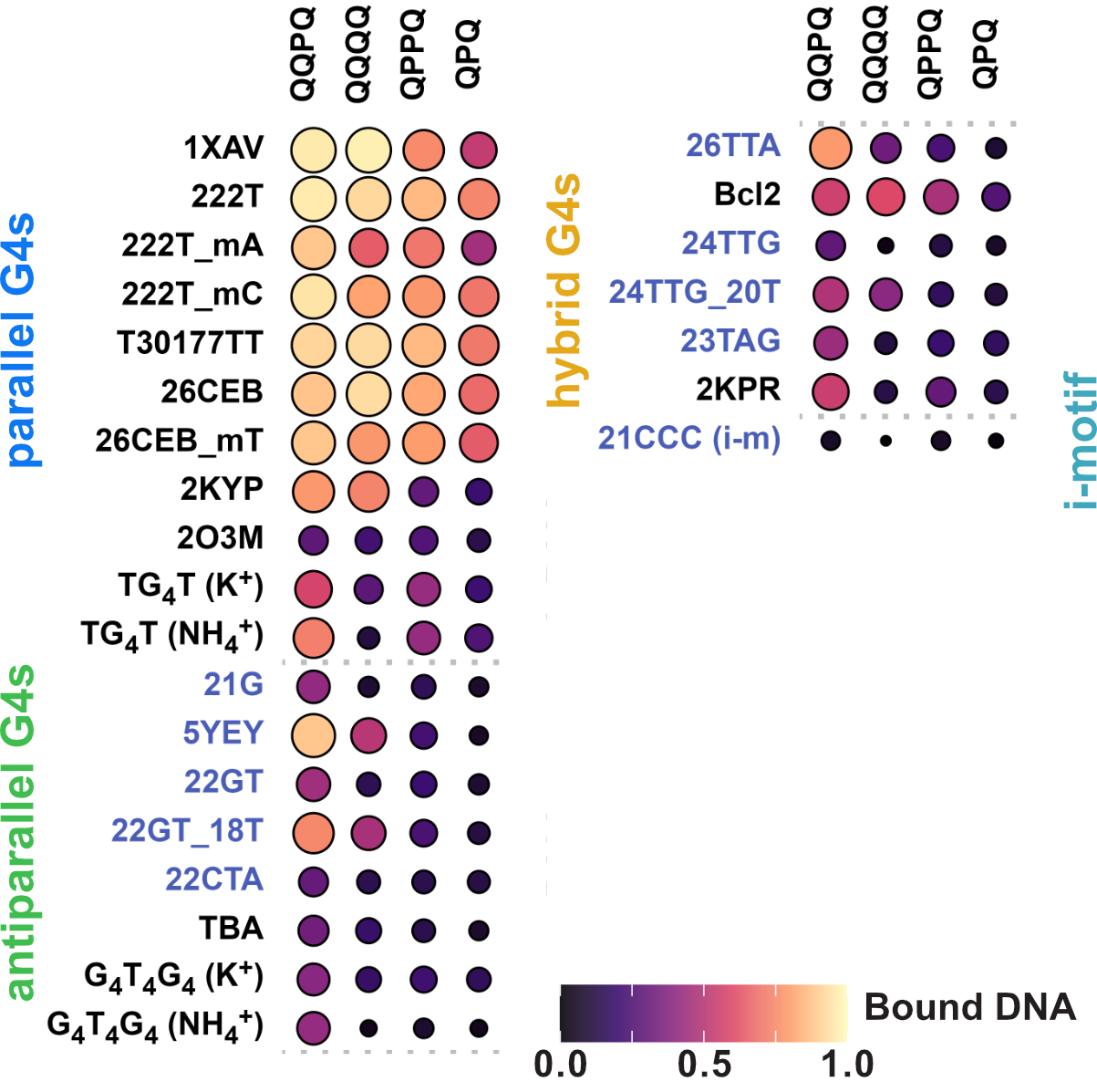
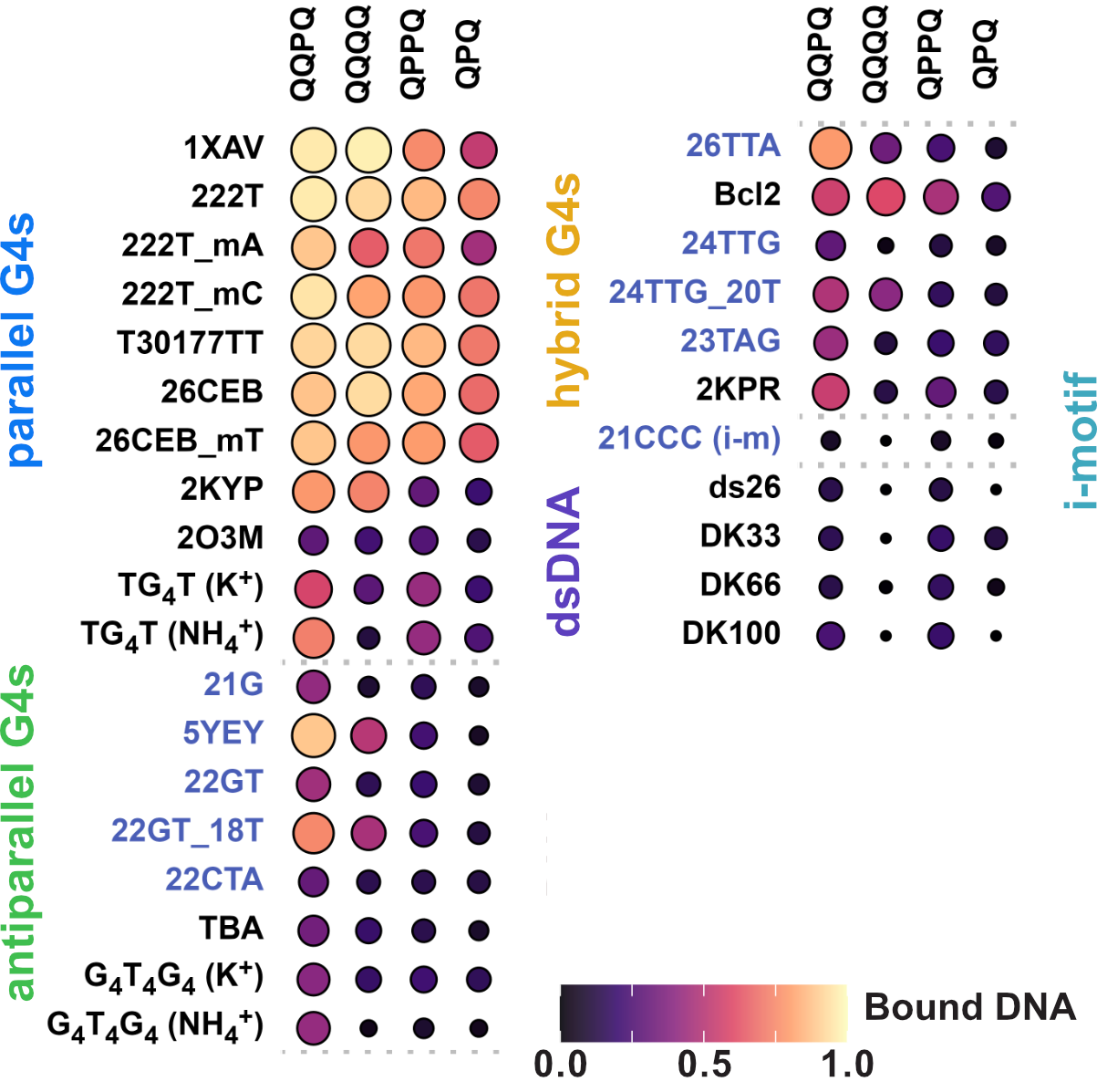
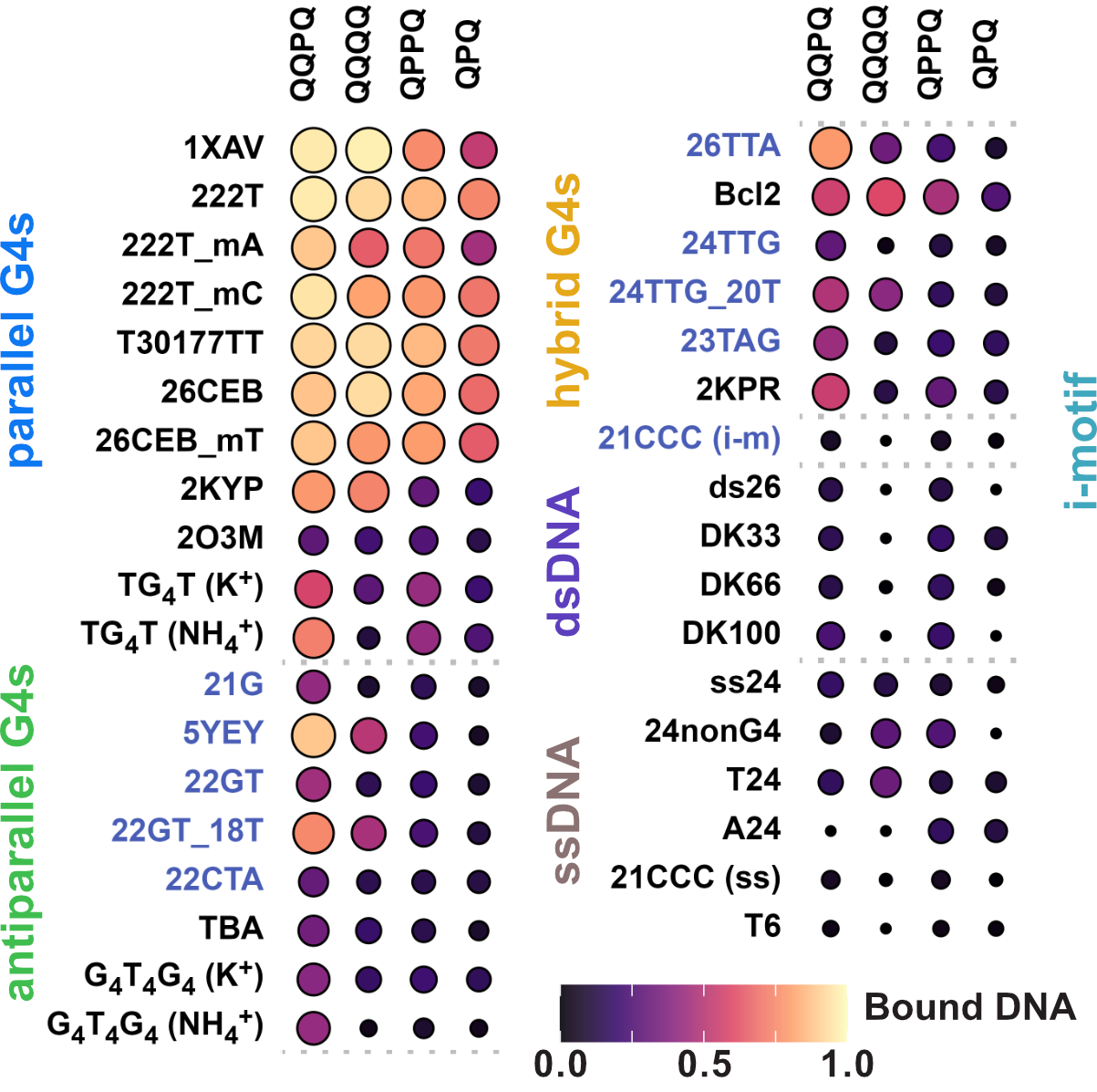
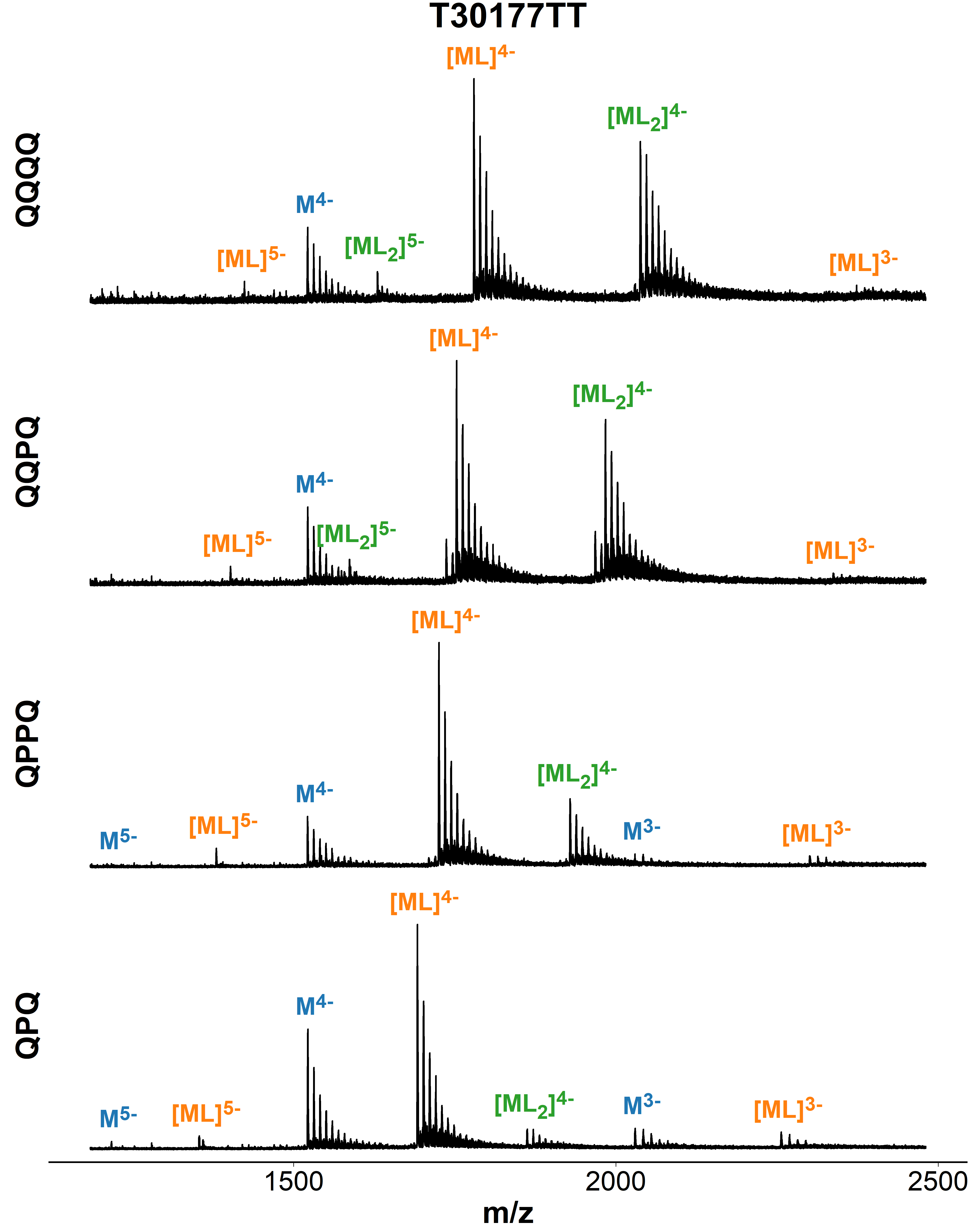
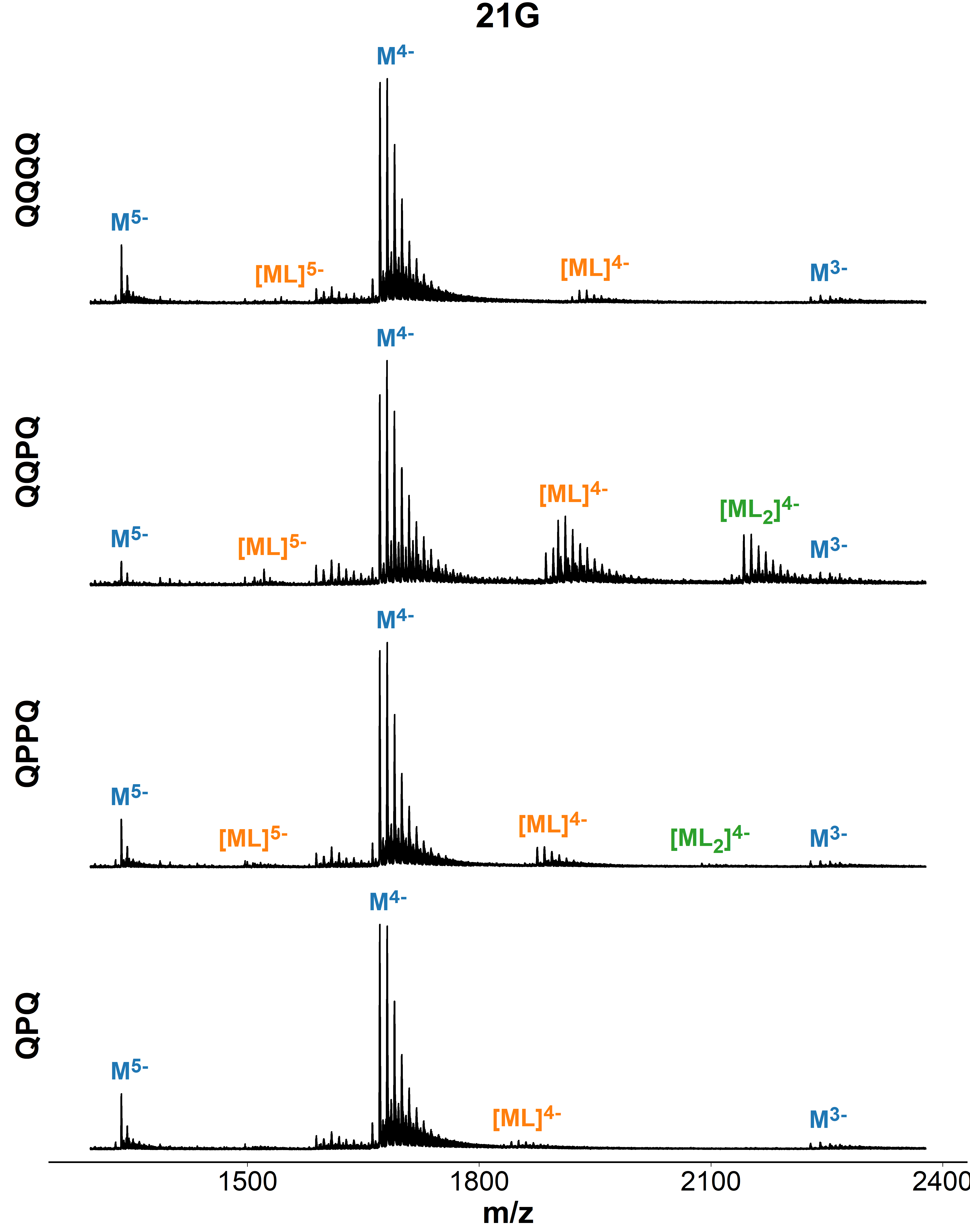
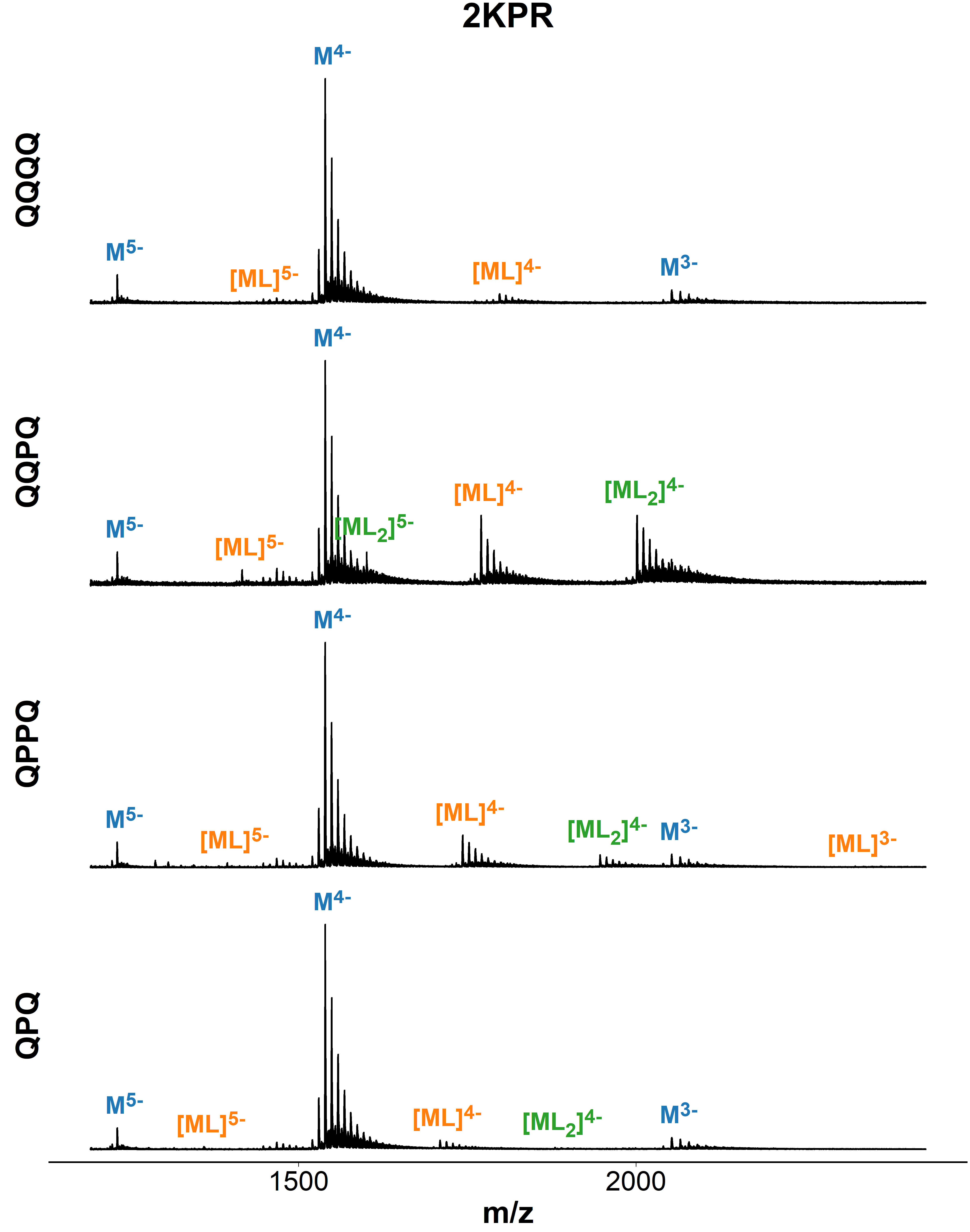
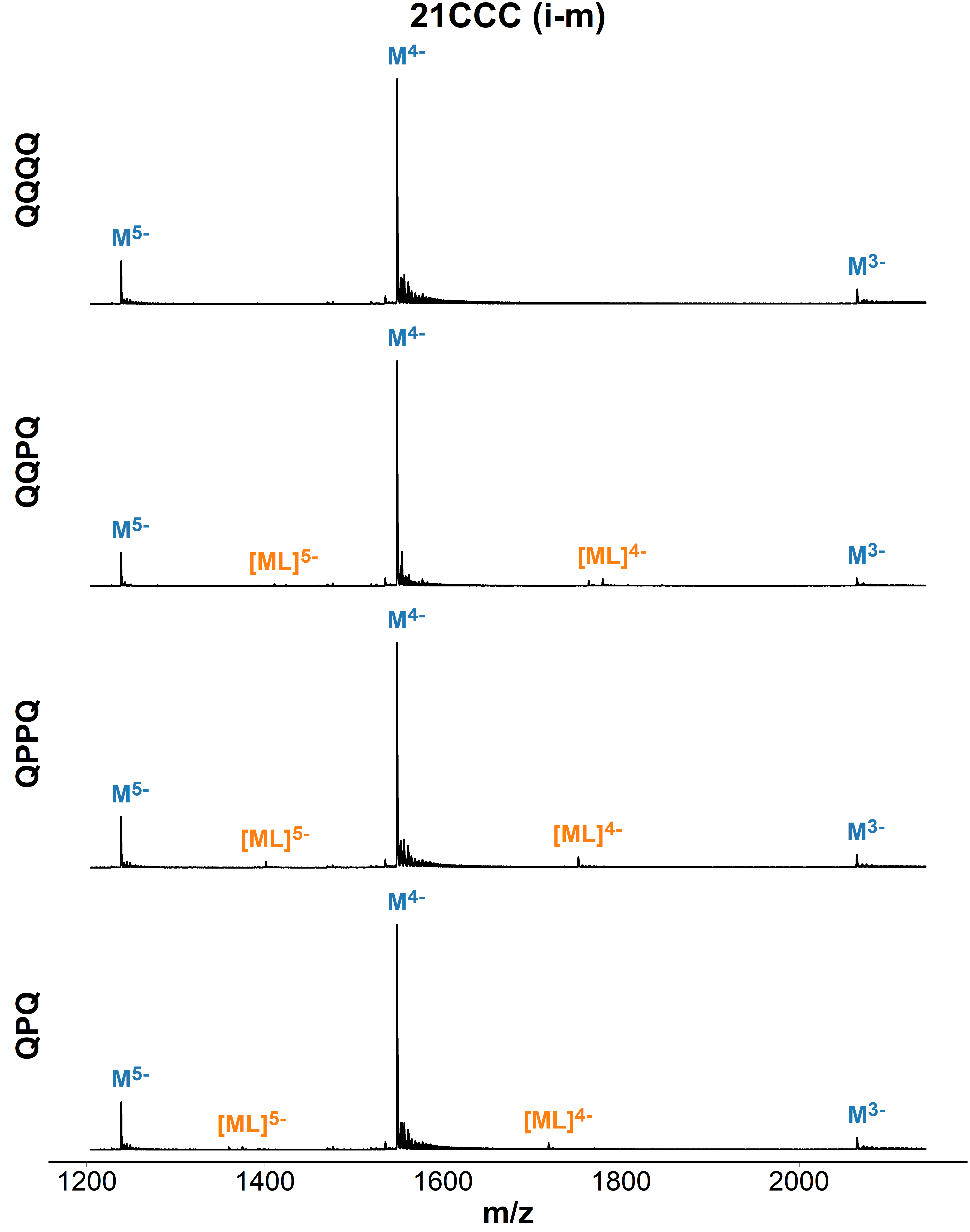
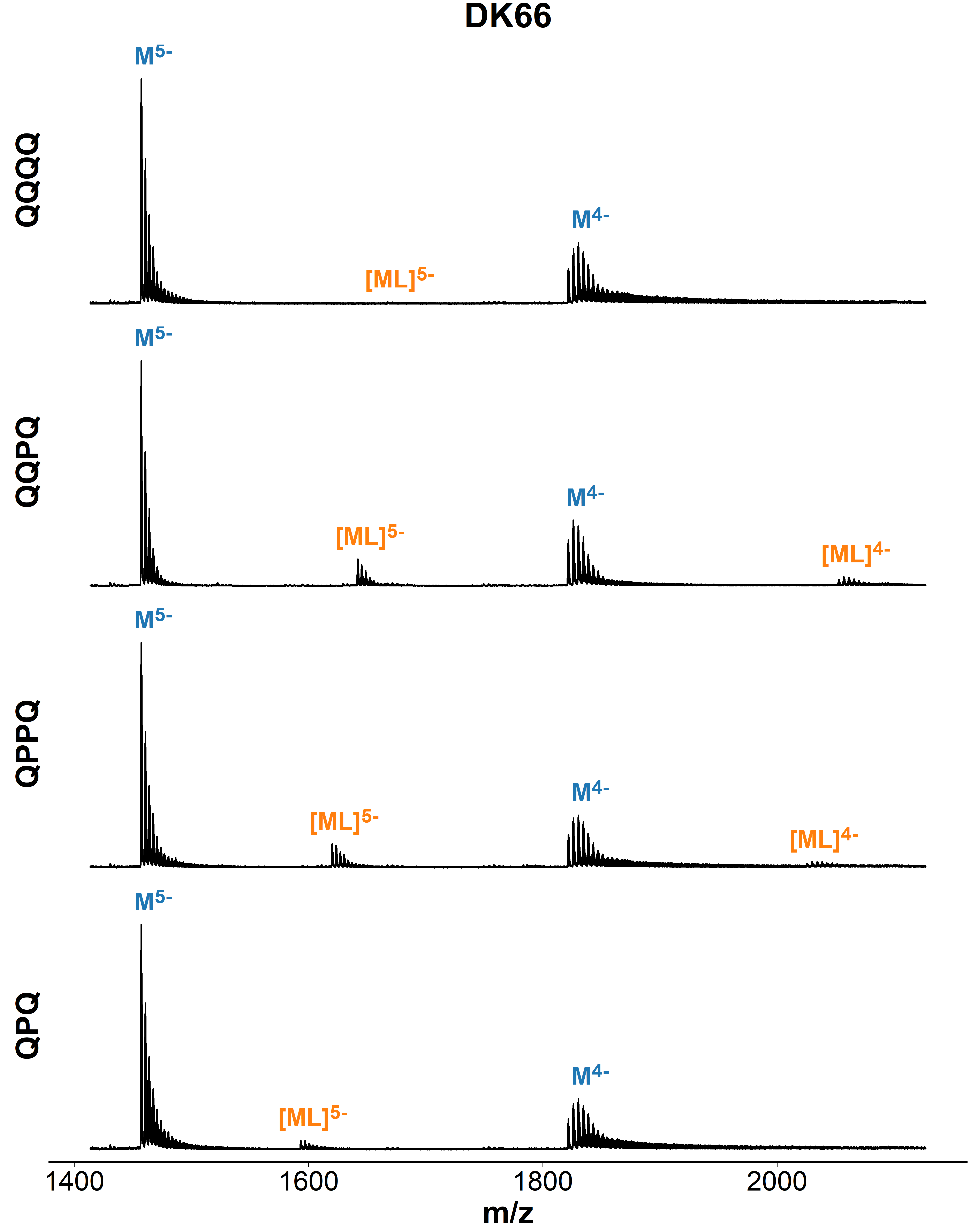

Selectivity confirmed by high-throughput nMS K titrations
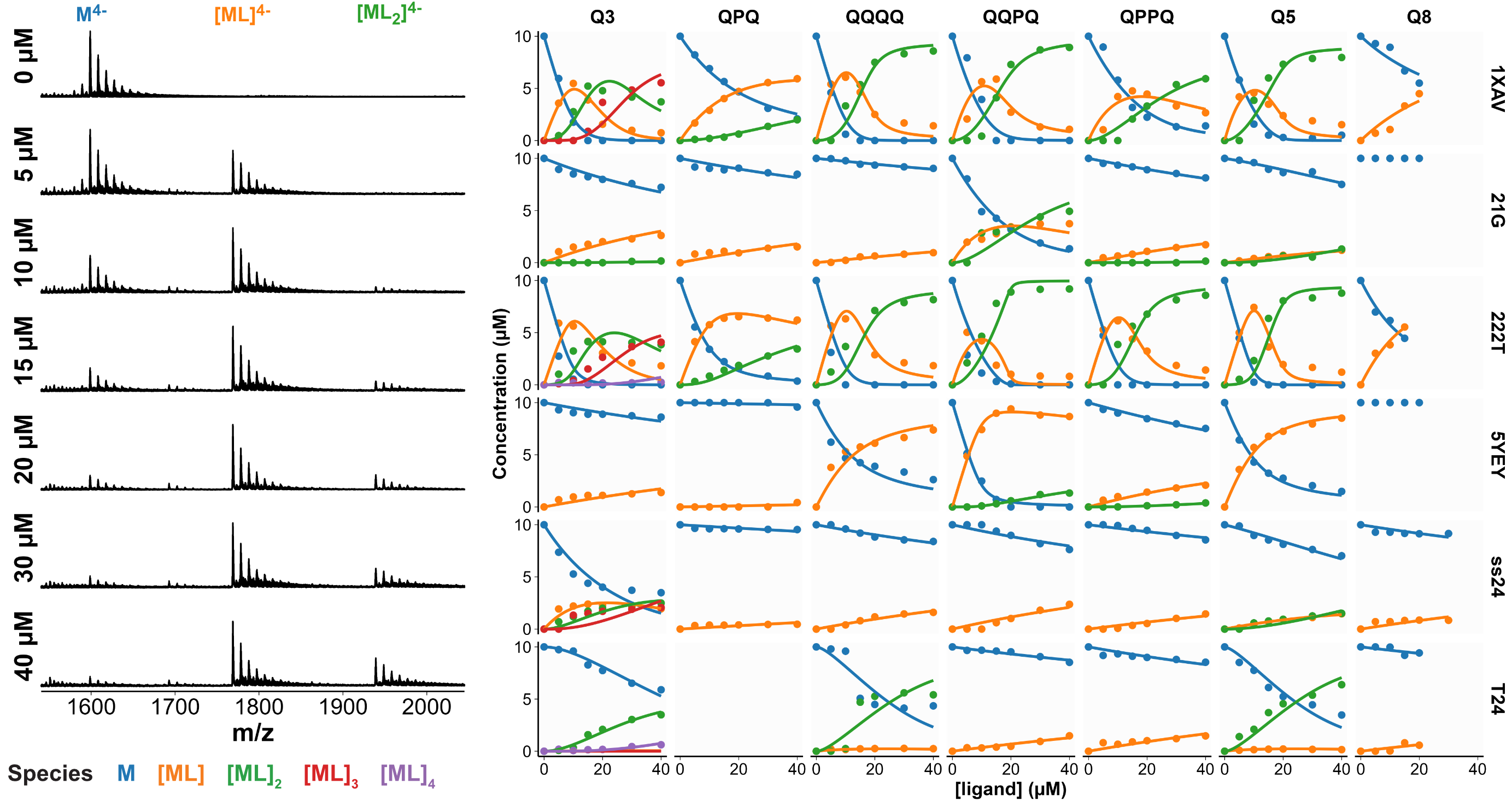
Not completely crystal clear binding
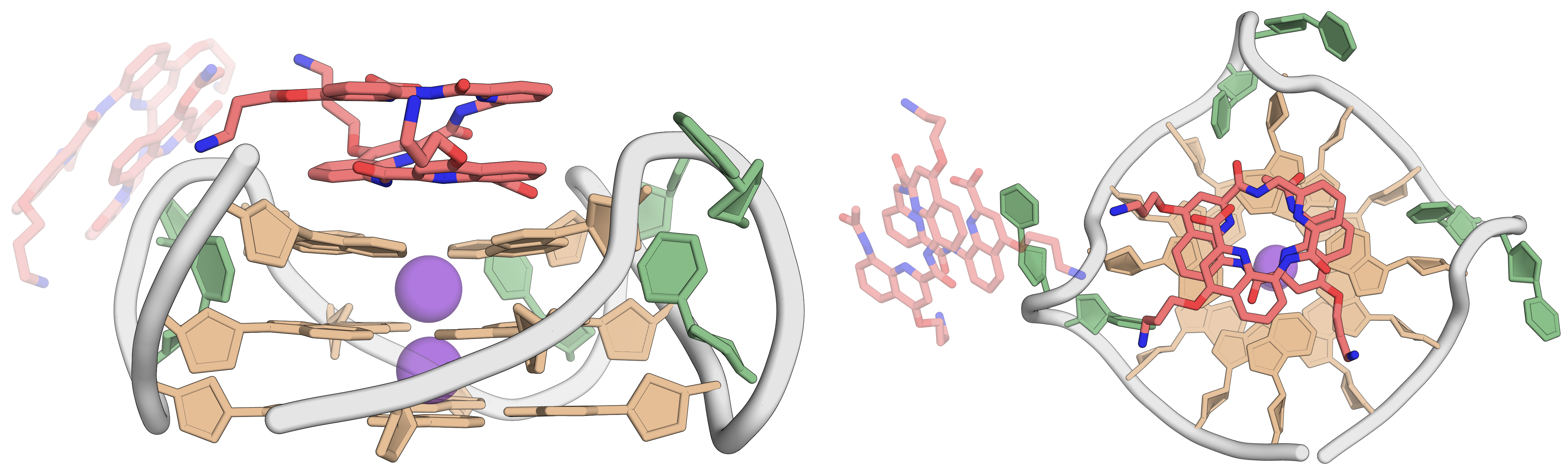

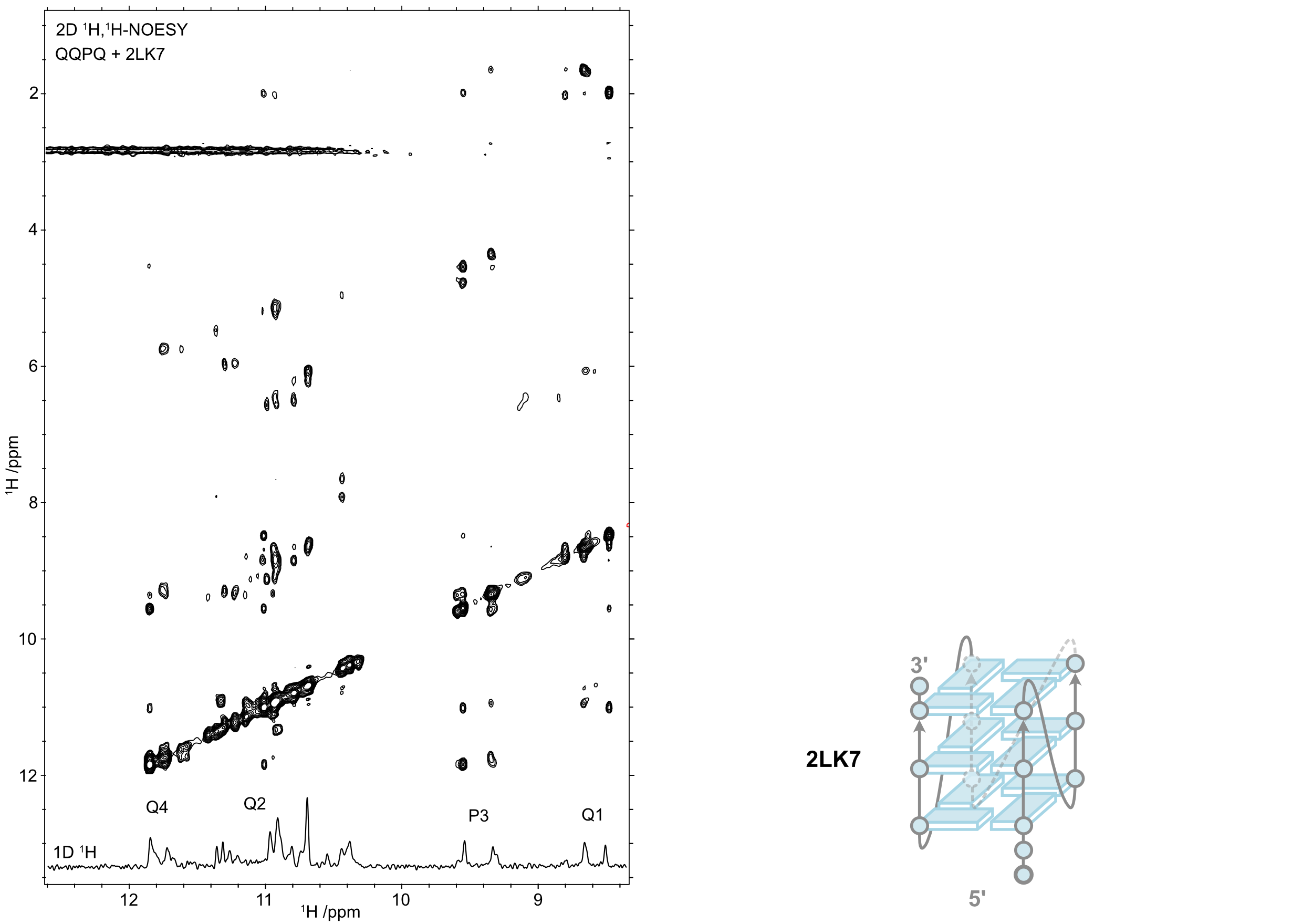
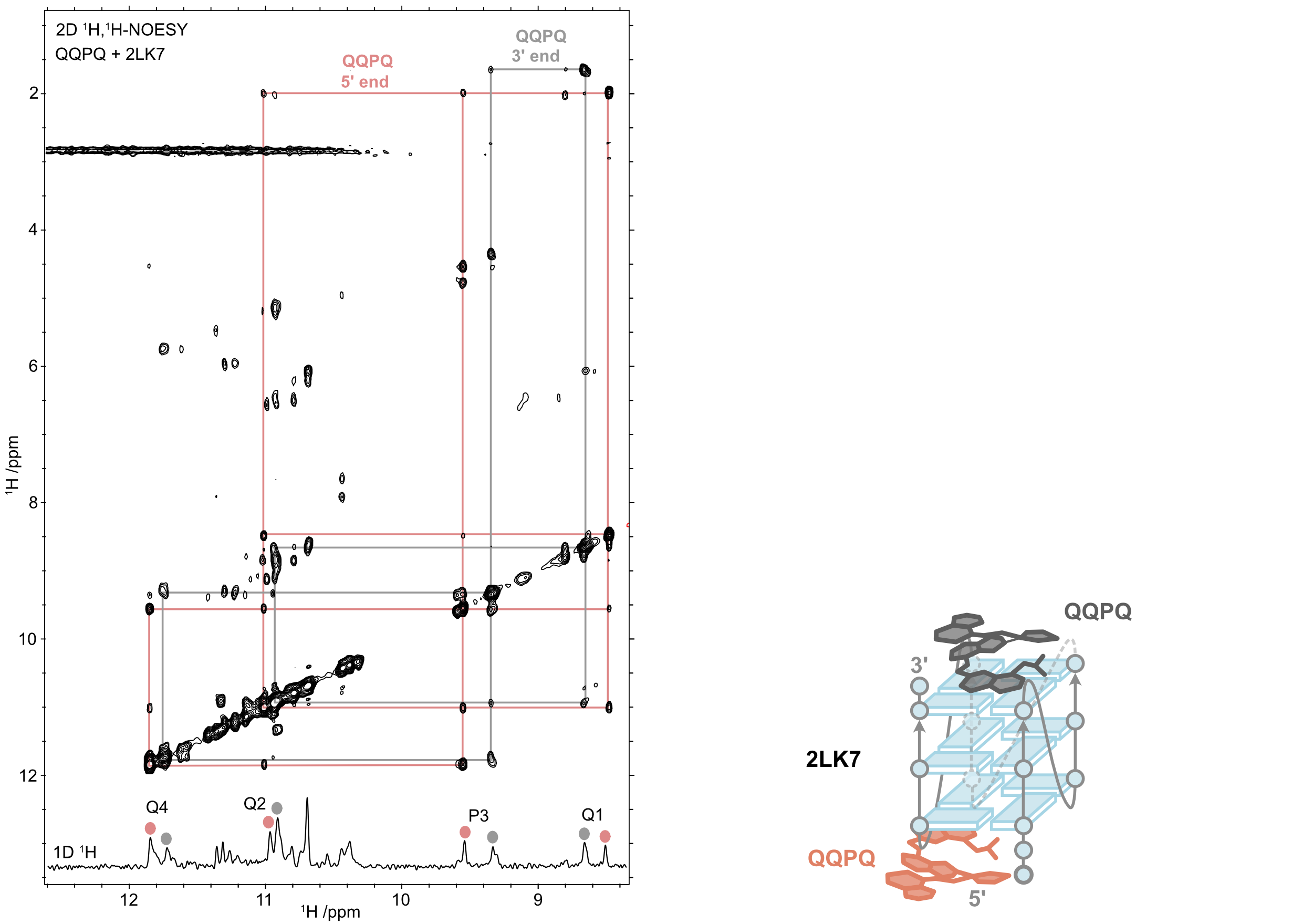
NMR confirms the stacking on both ends
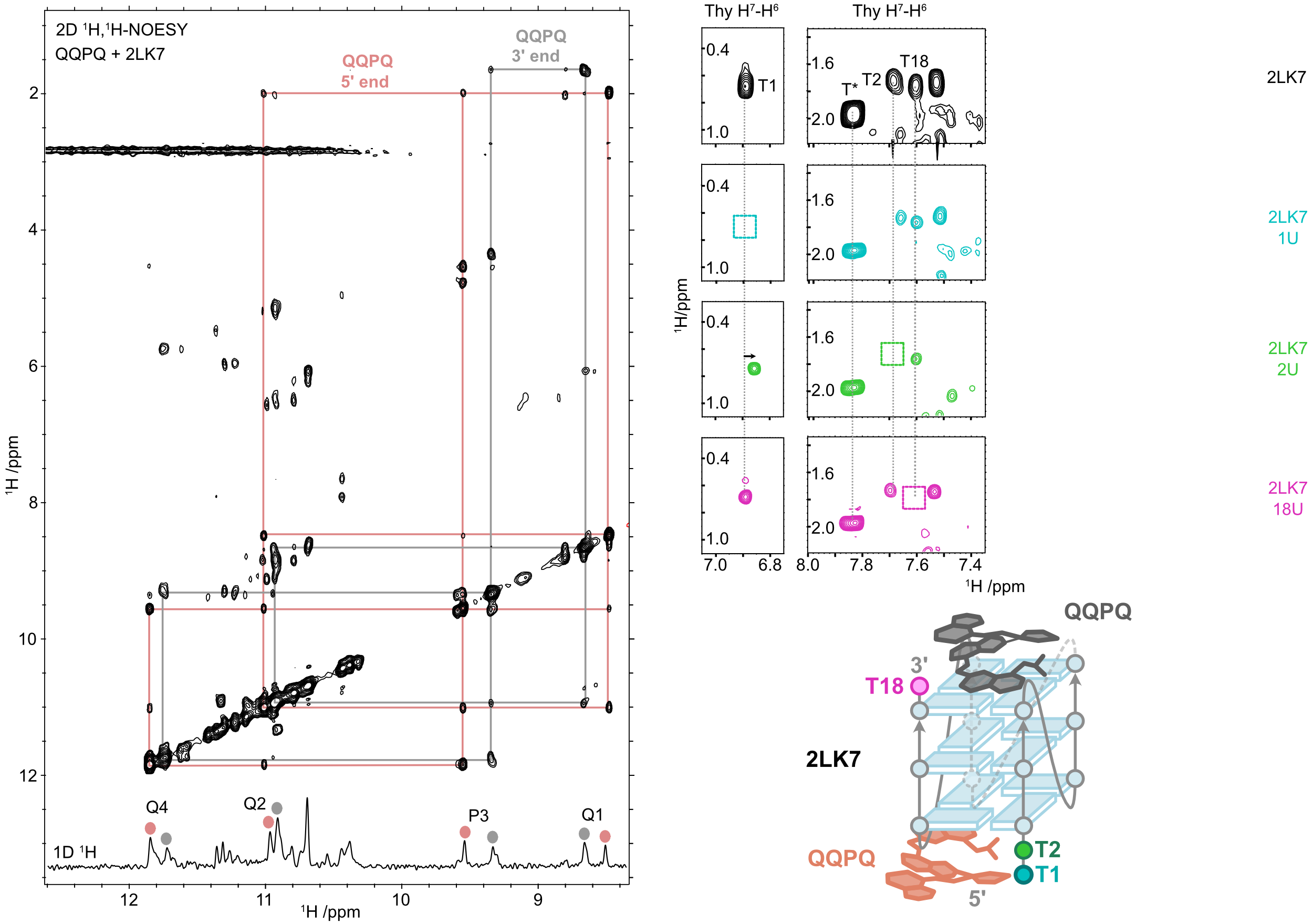
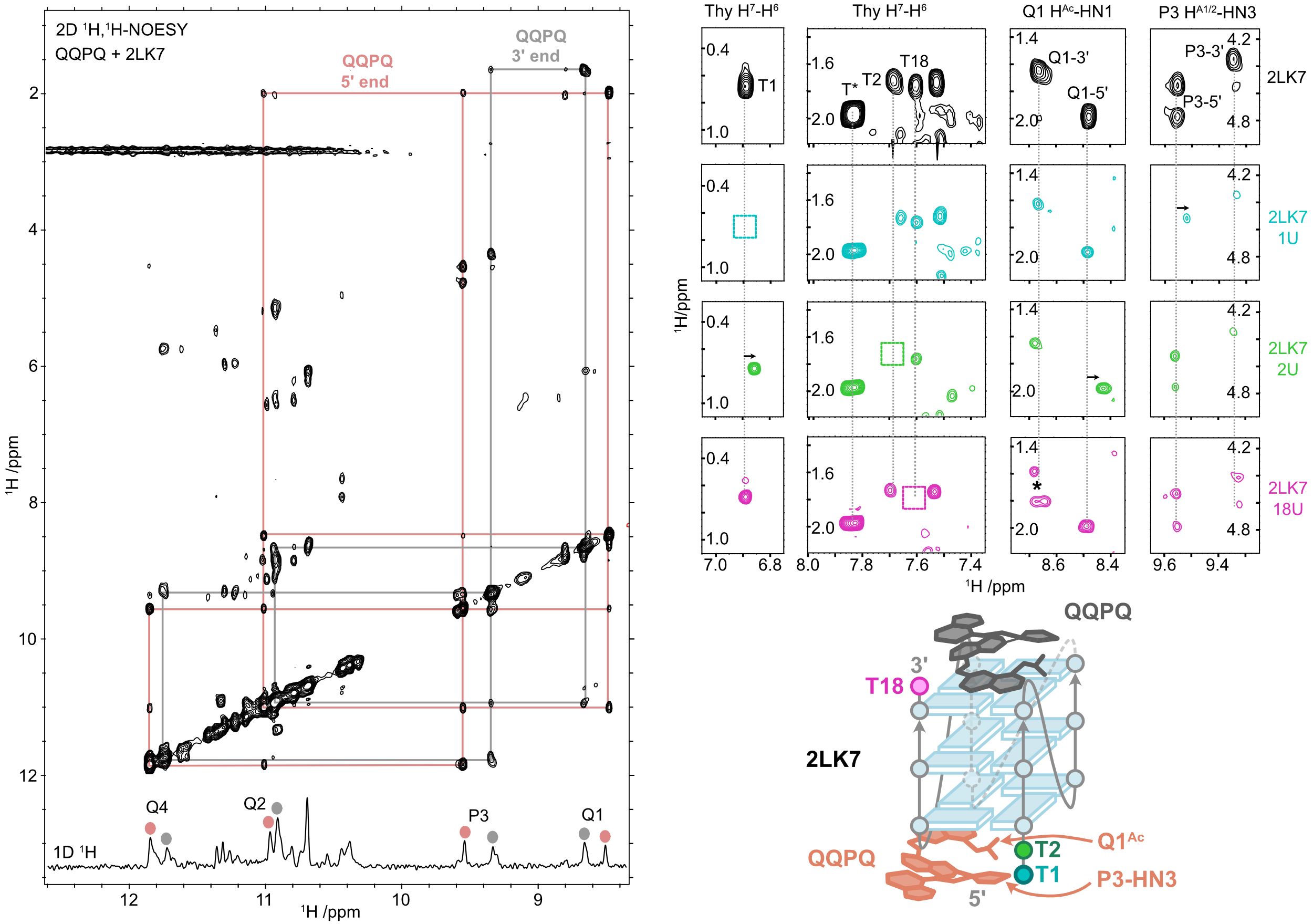

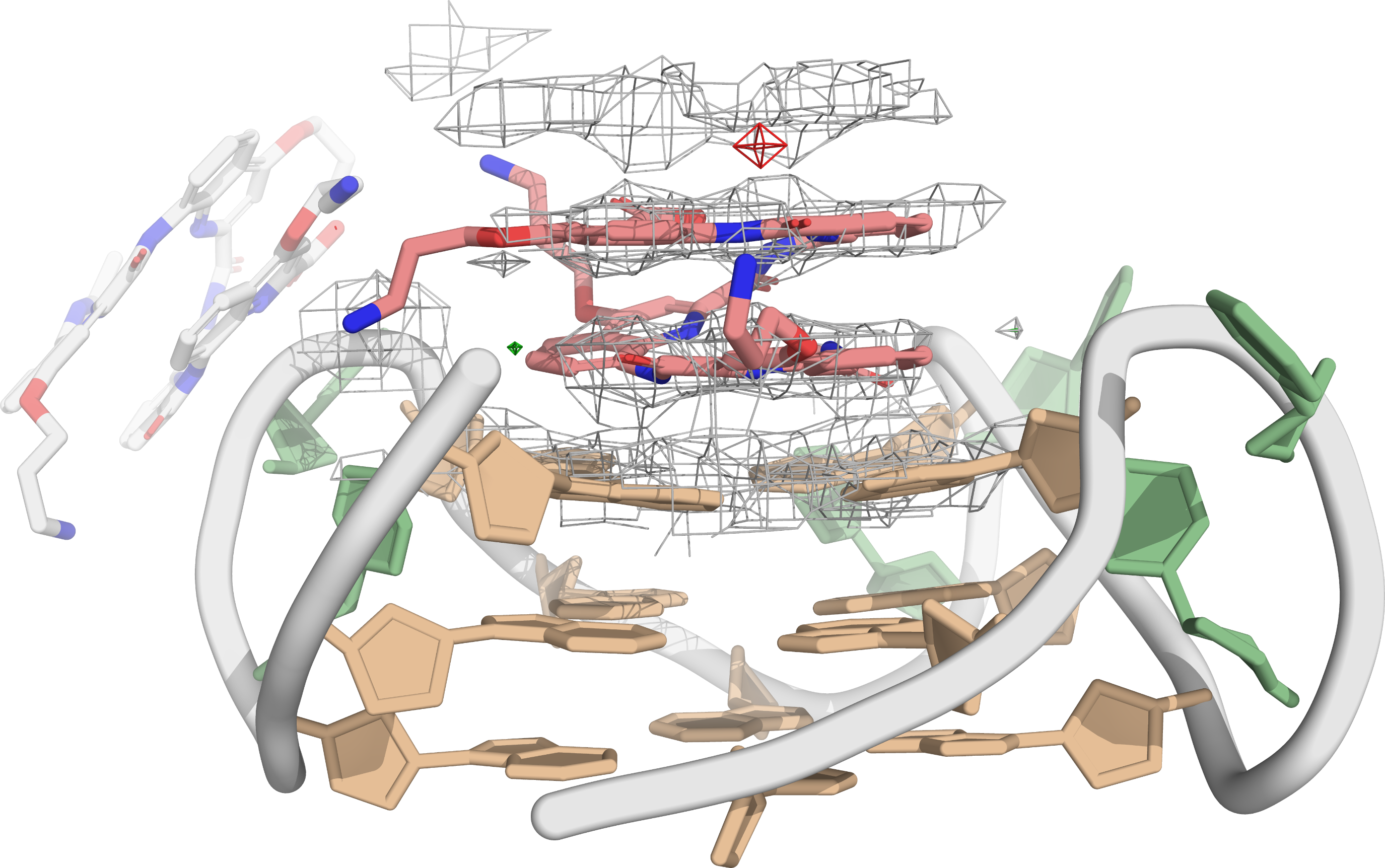
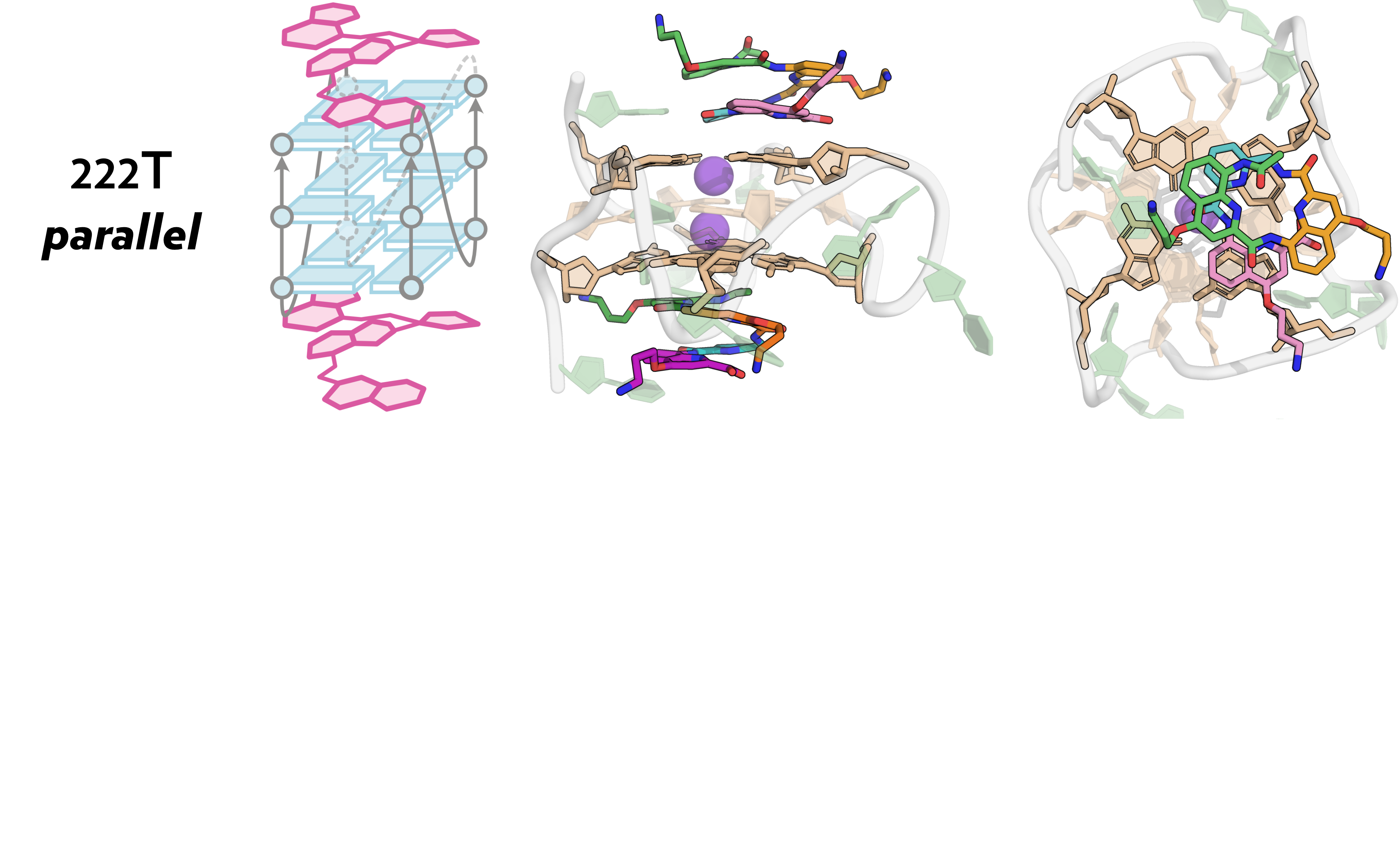
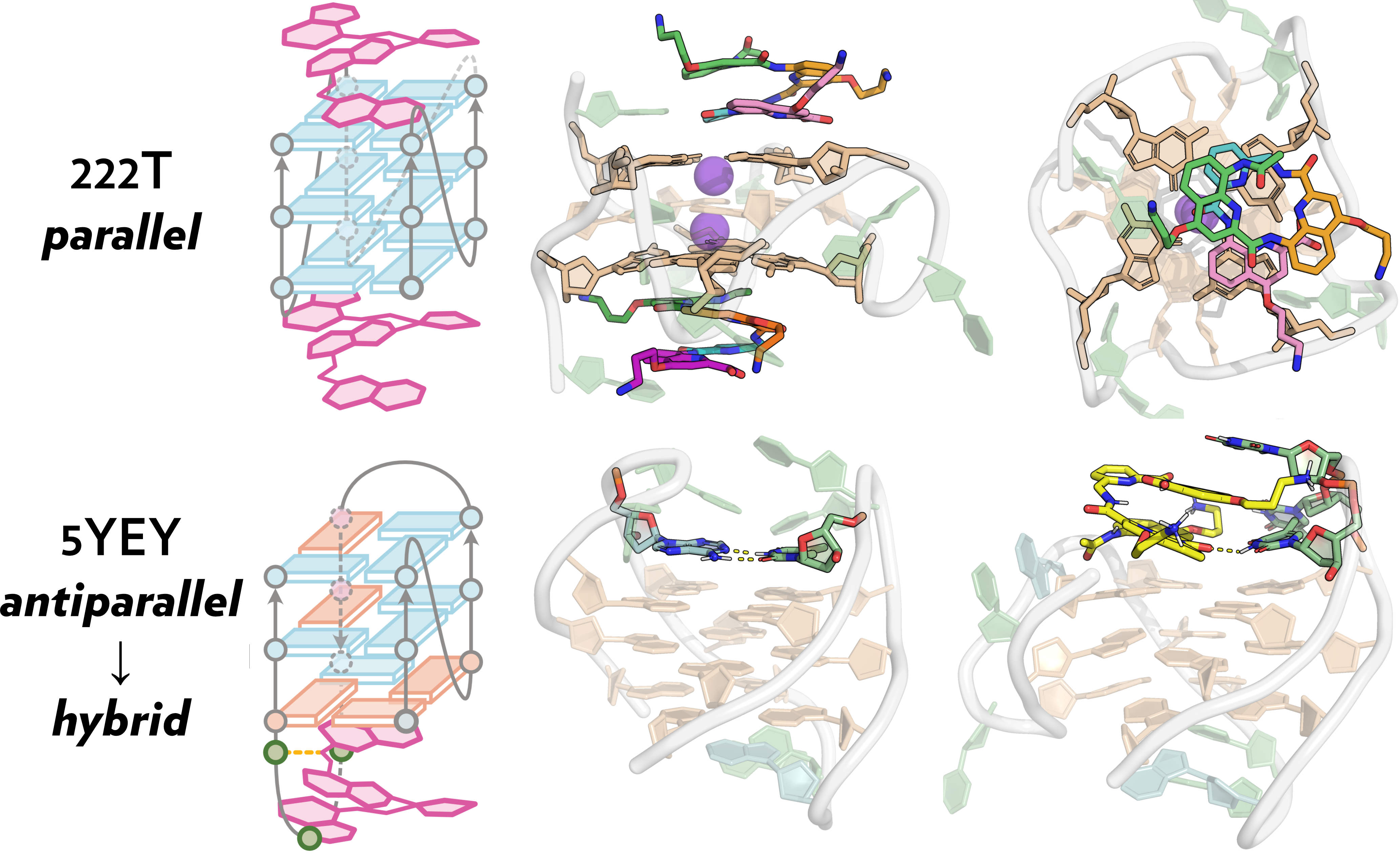
Final models supported by MD simulations
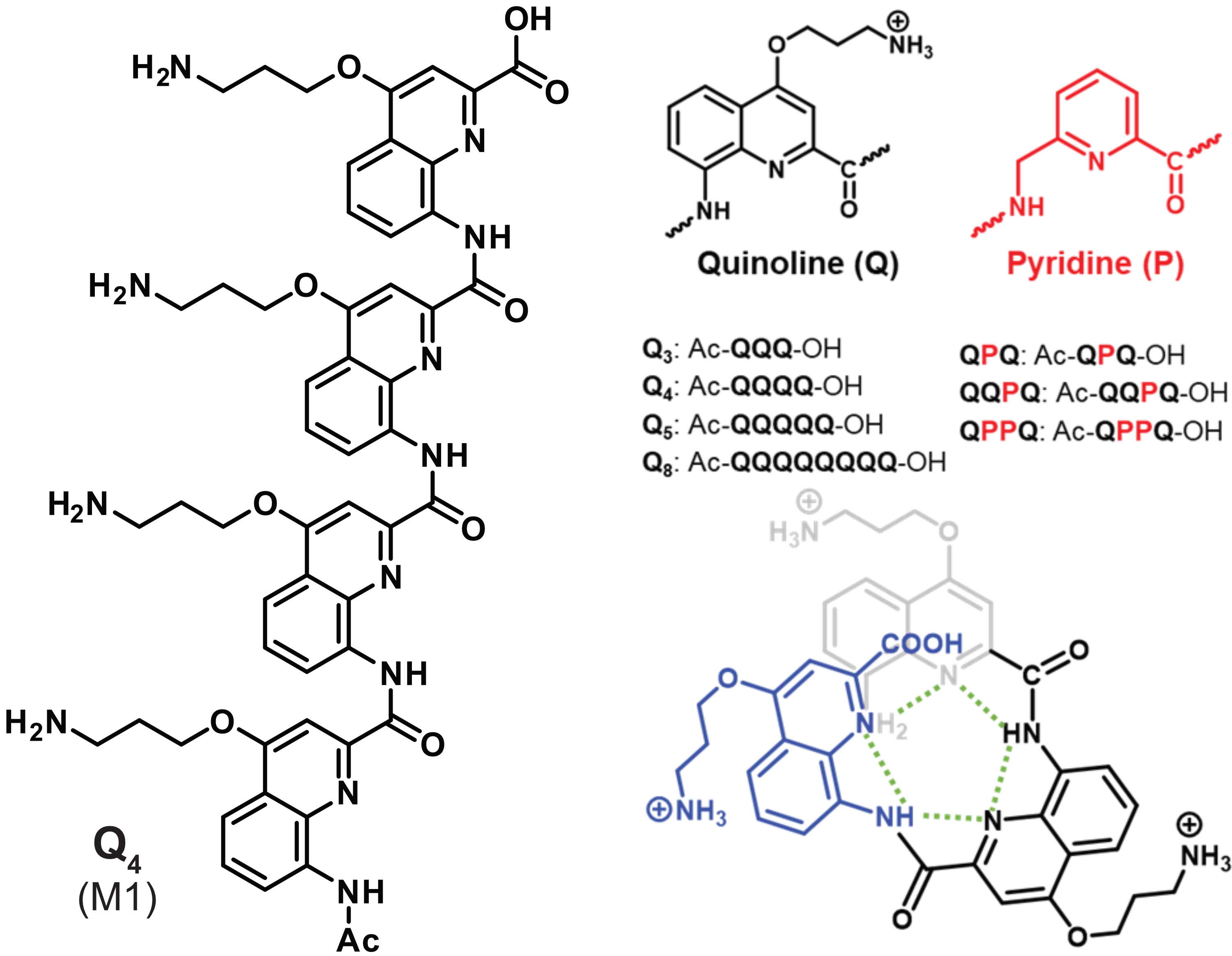
Foldamers allow precisal structural control
of artifical protein assemblies
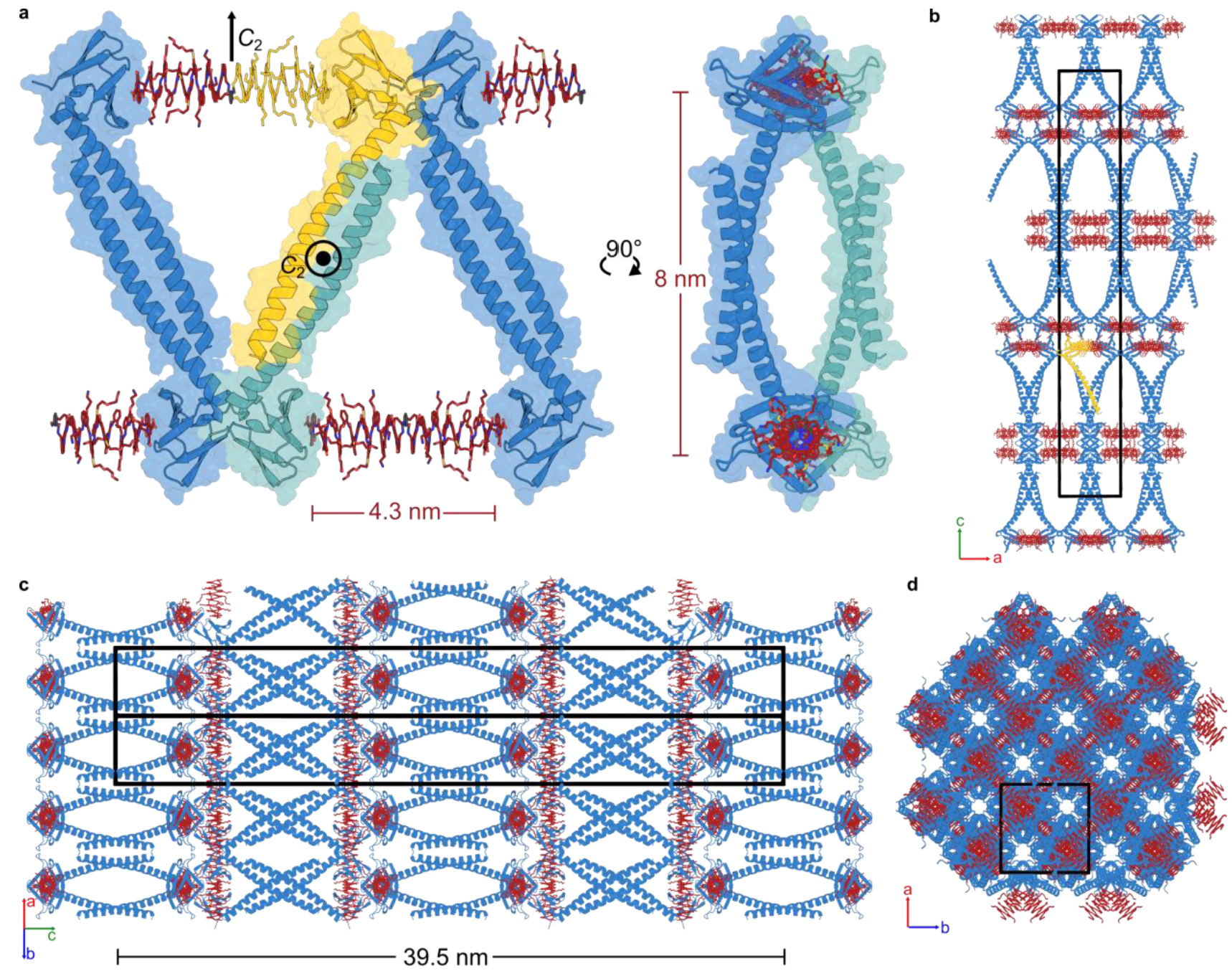

SEC and nMS are methods of choice
to determine stoichiometries
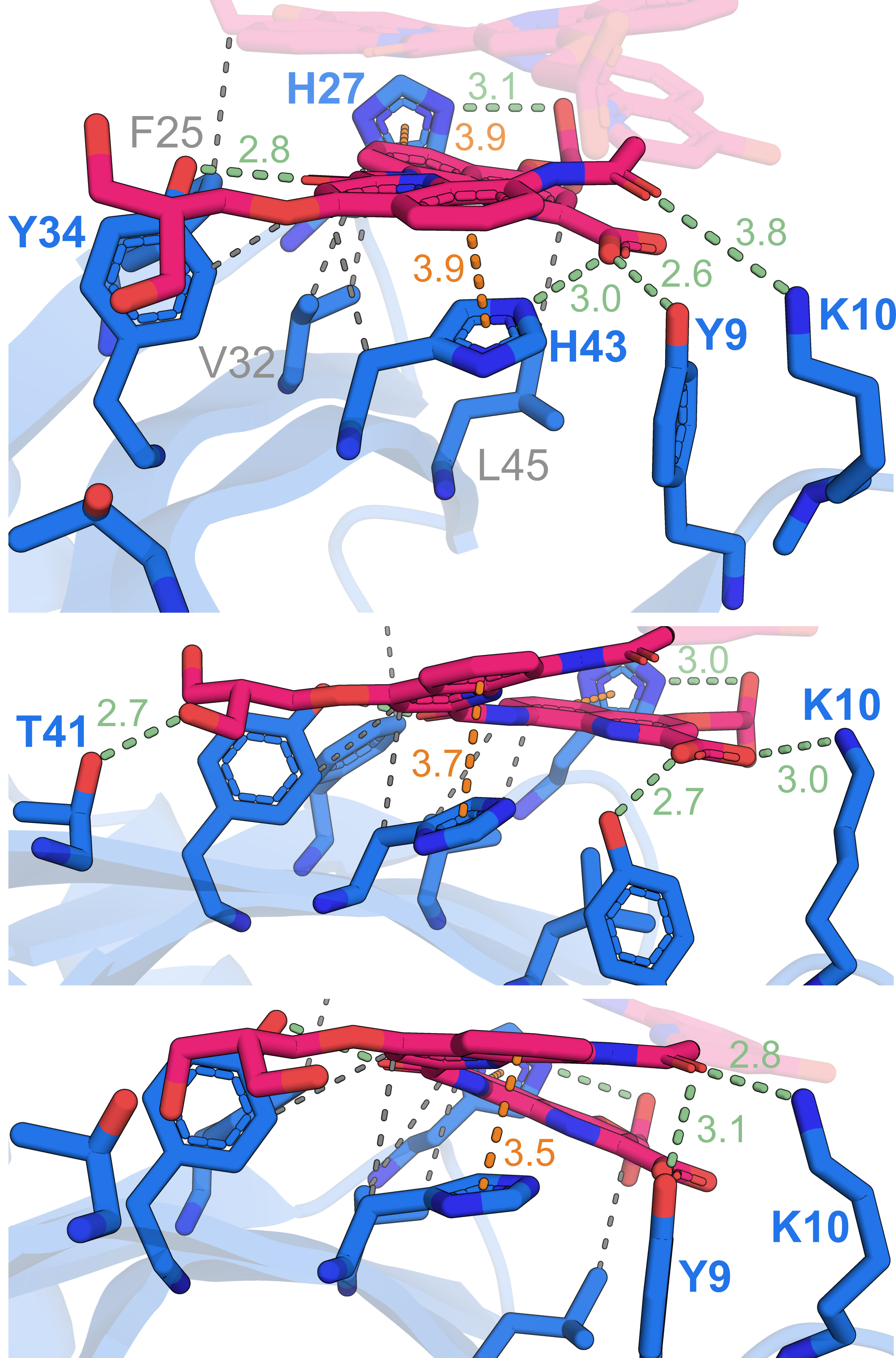
Foldamers also bind proteins in unexpected ways
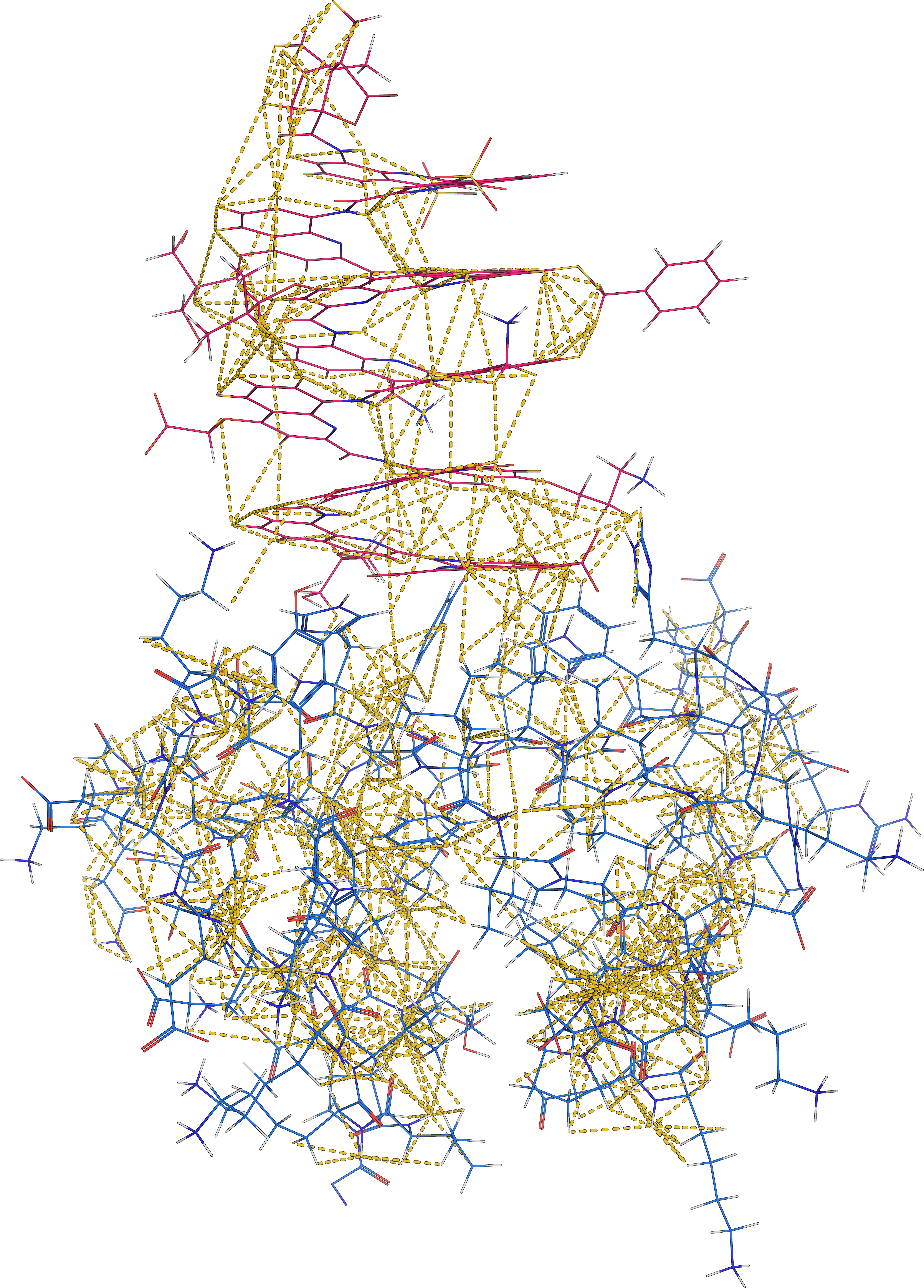

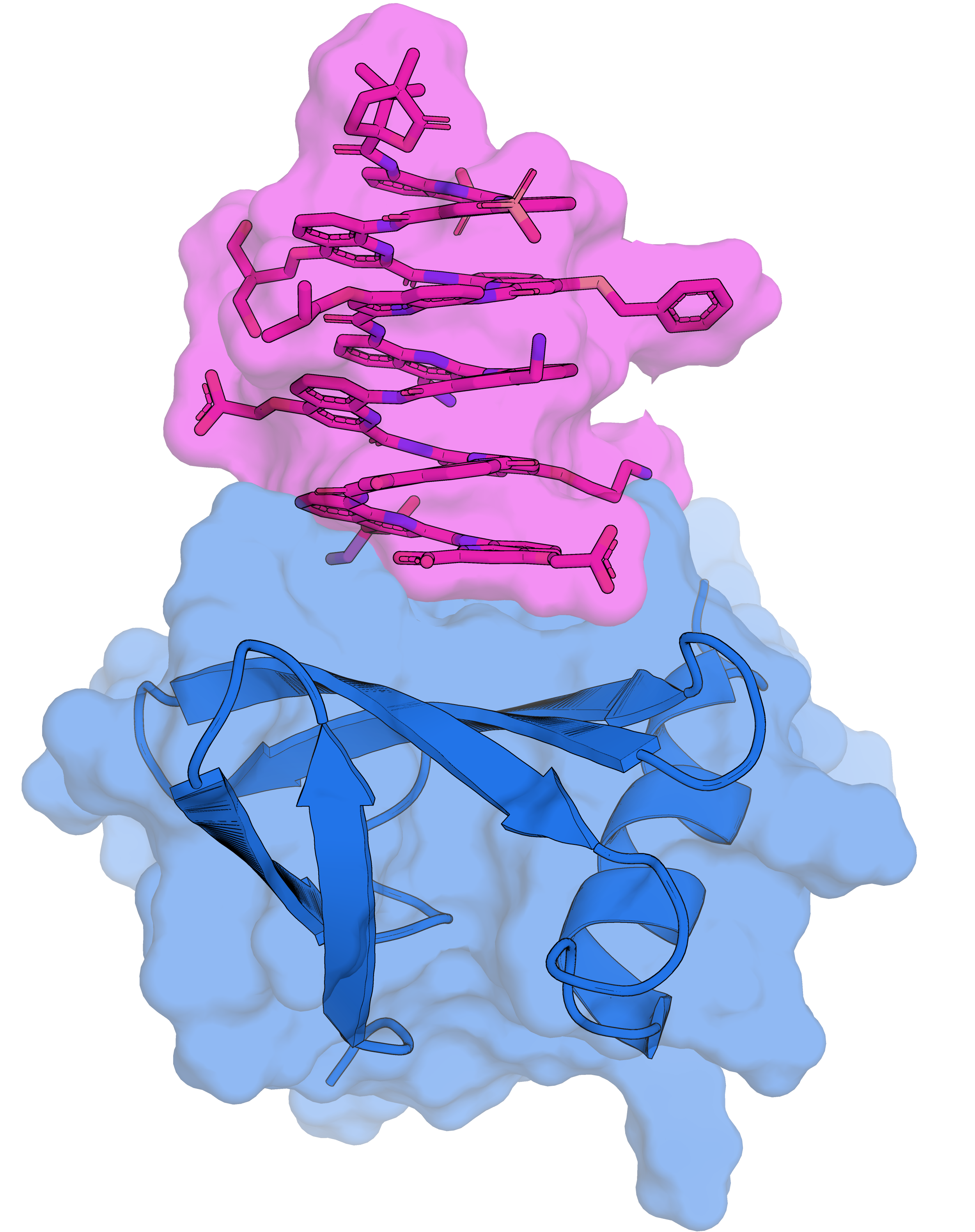

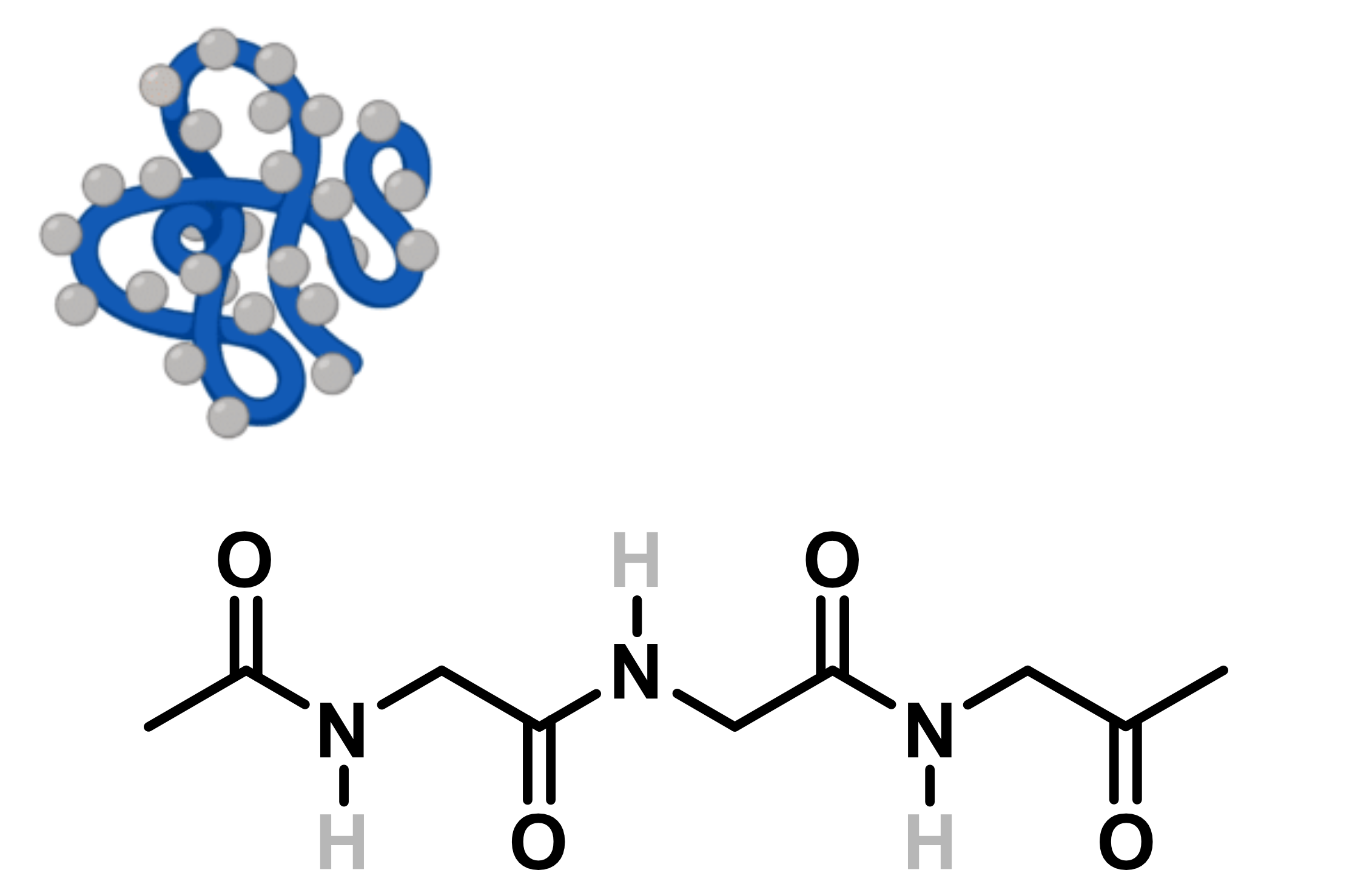
HDX/MS gives insights
into protein structural dynamics

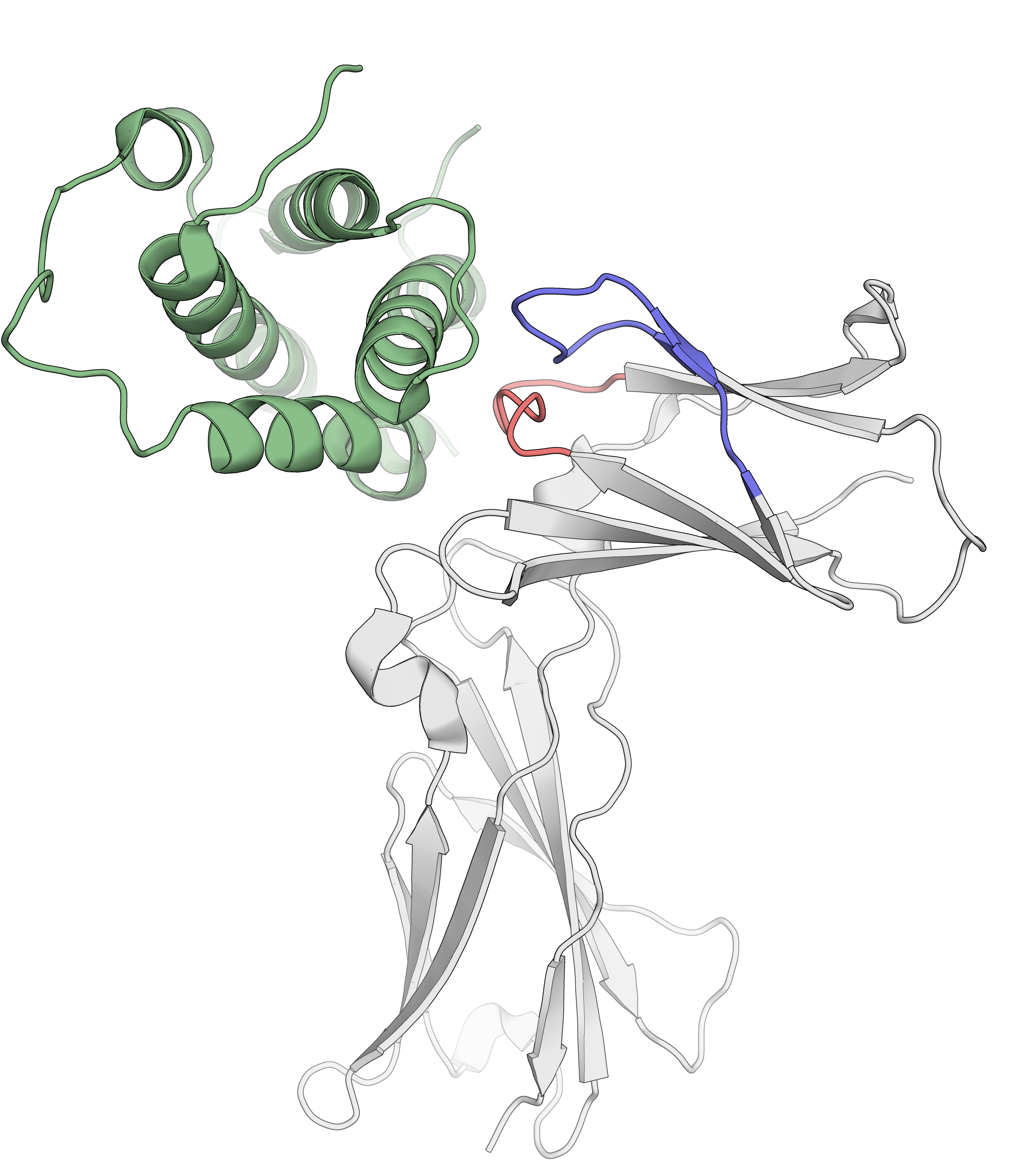
HDX/MS allows epitope mapping
in regulated environments
![]()
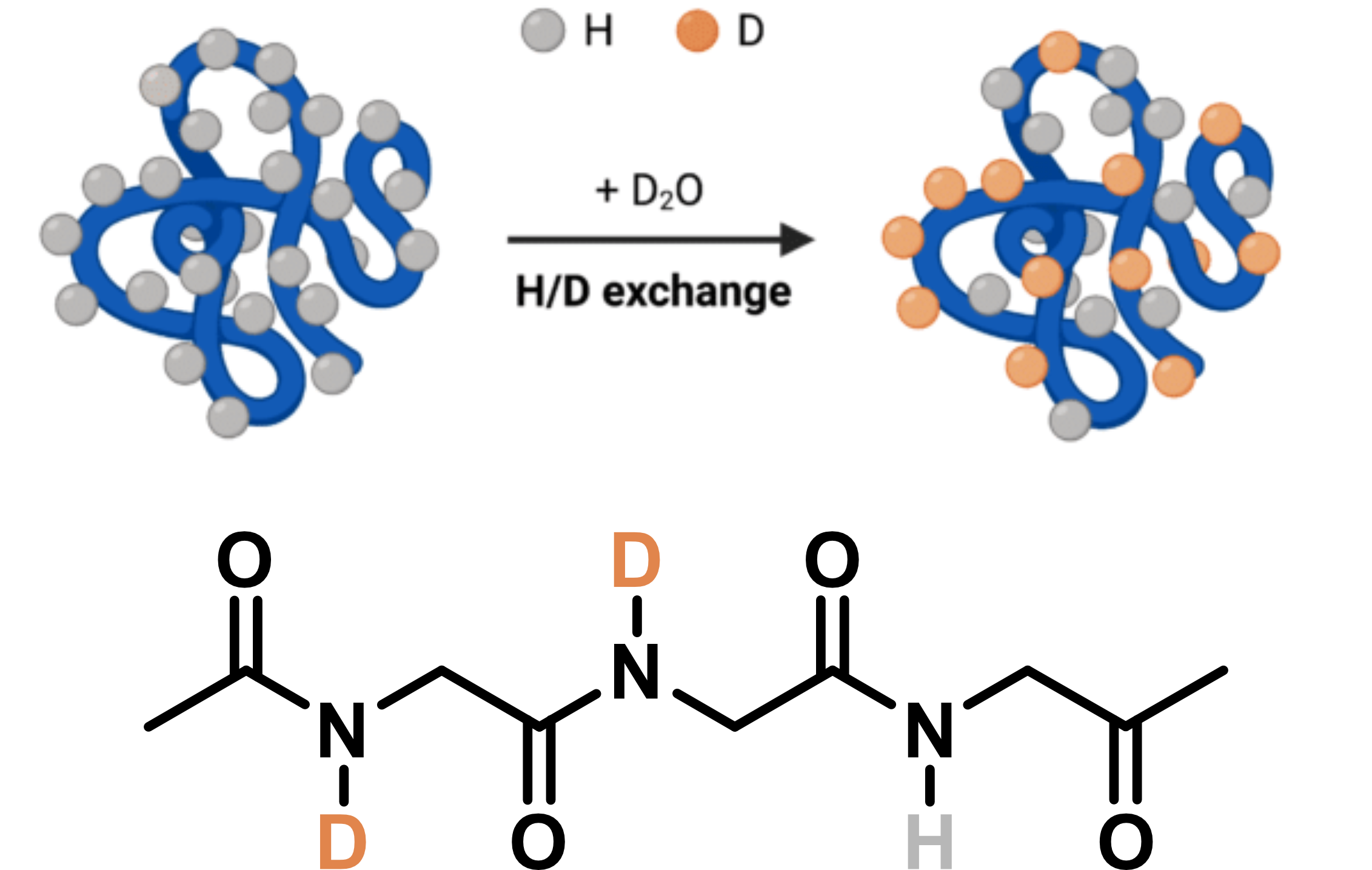
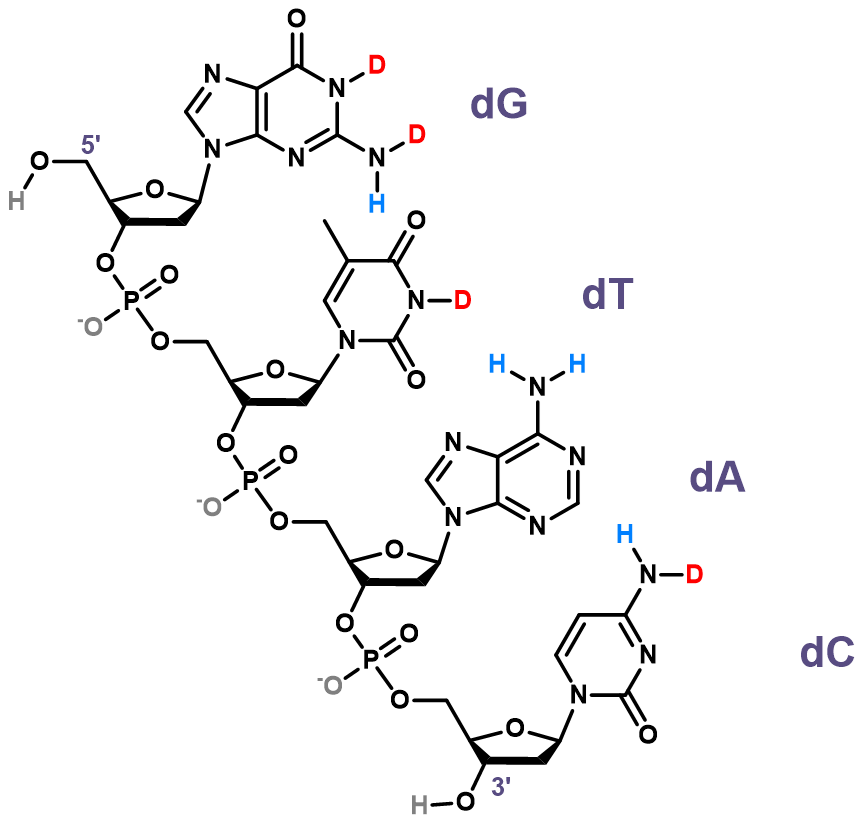
DNA is amenable to HDX/MS
- H/D exchange ideal for structure probing
- Full sequence coverage
- Directly involved in folding
- Minimal primary/secondary structure change
- Rate of HDX should be structure dependent
- Native MS measurements

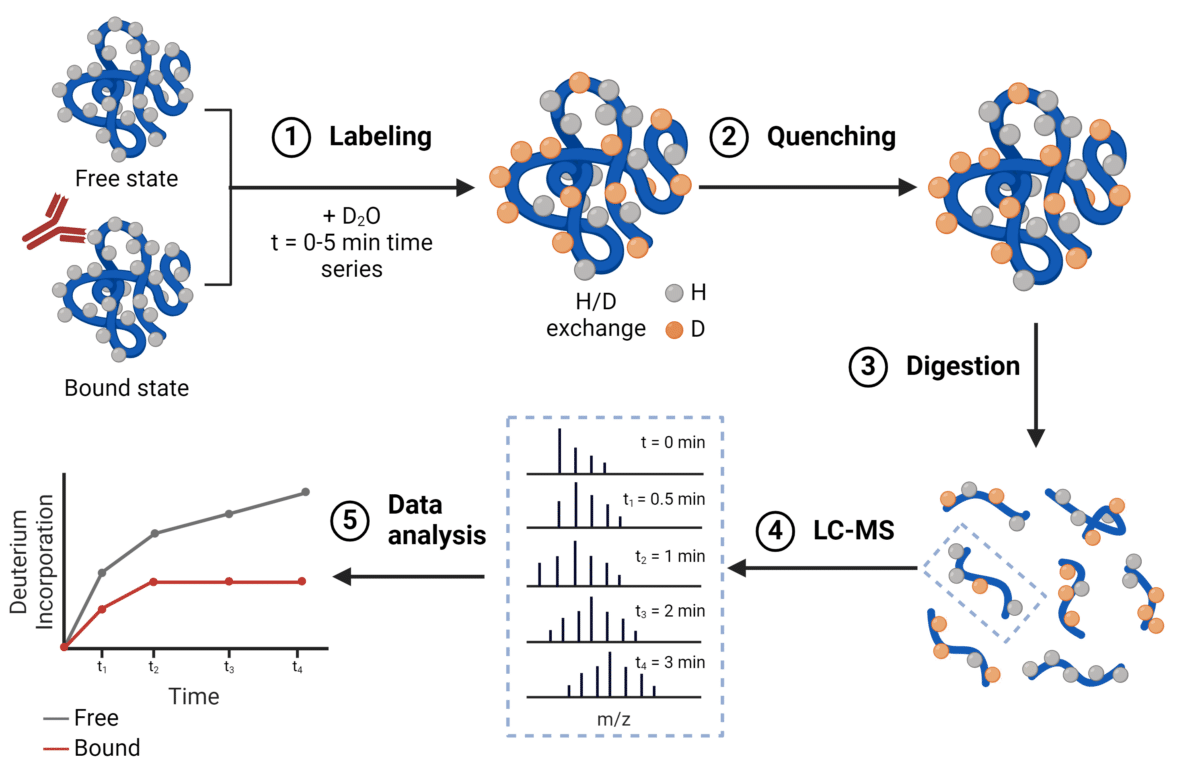
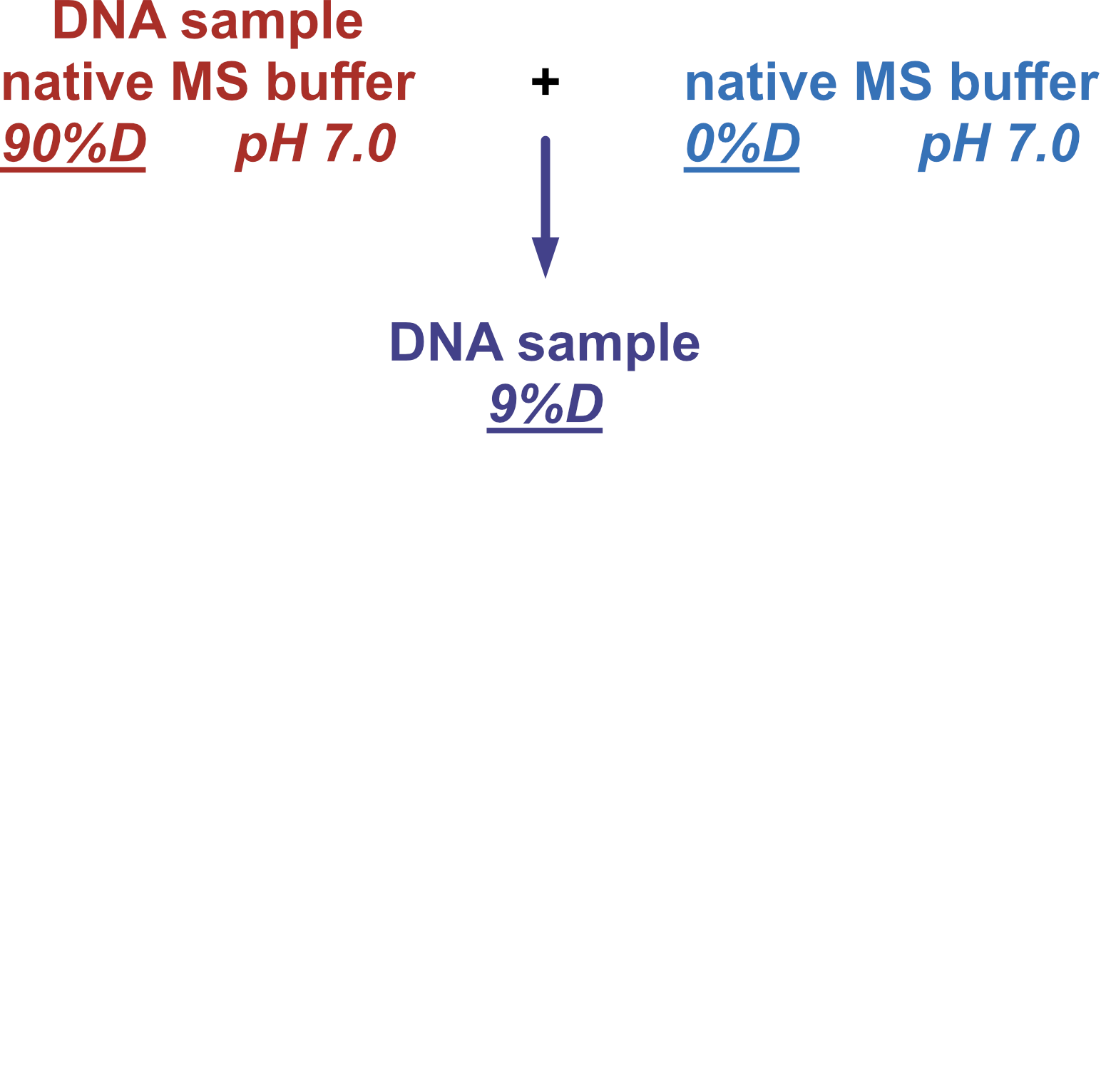
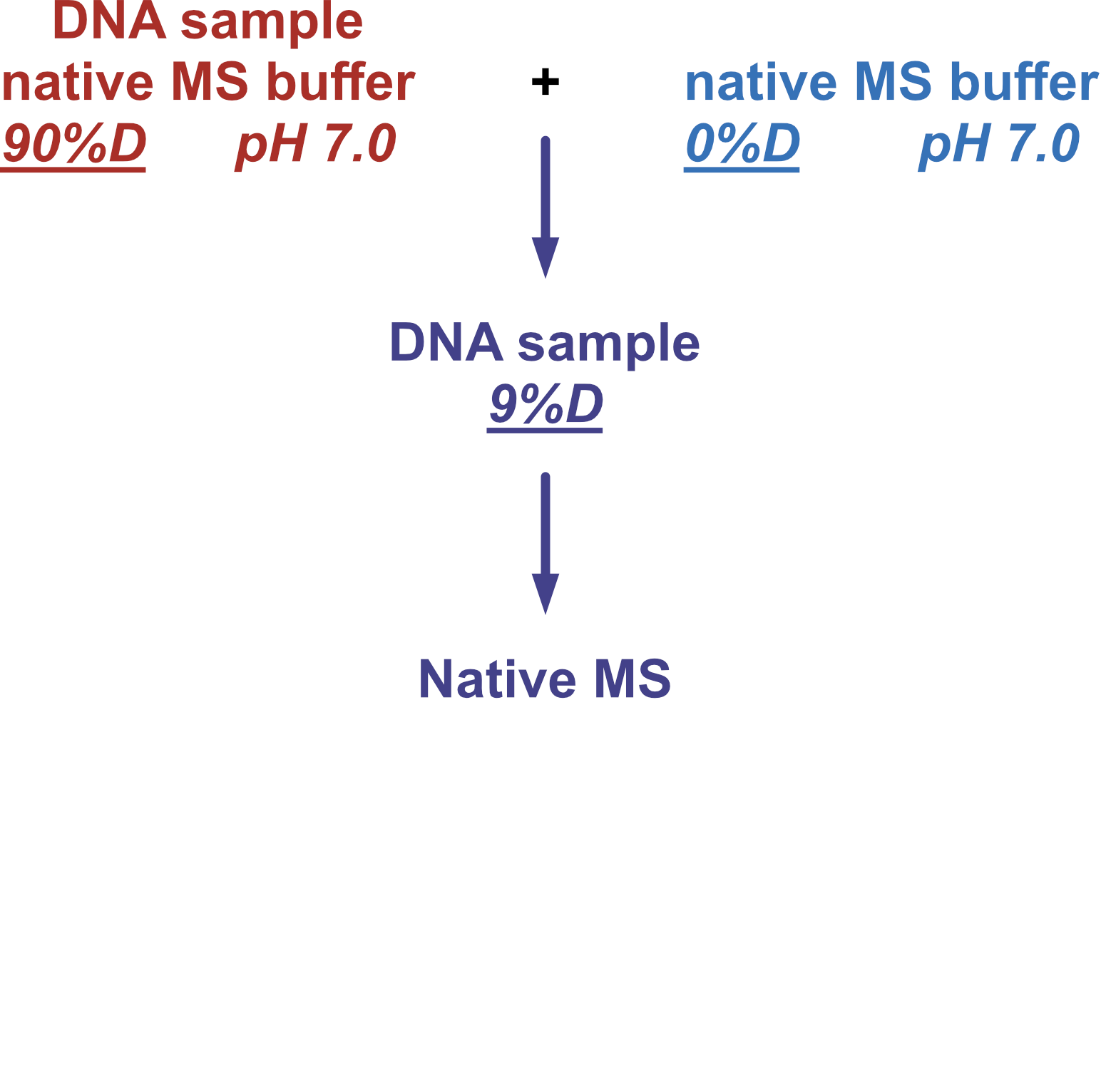
Coupling in-solution HDX to native MS

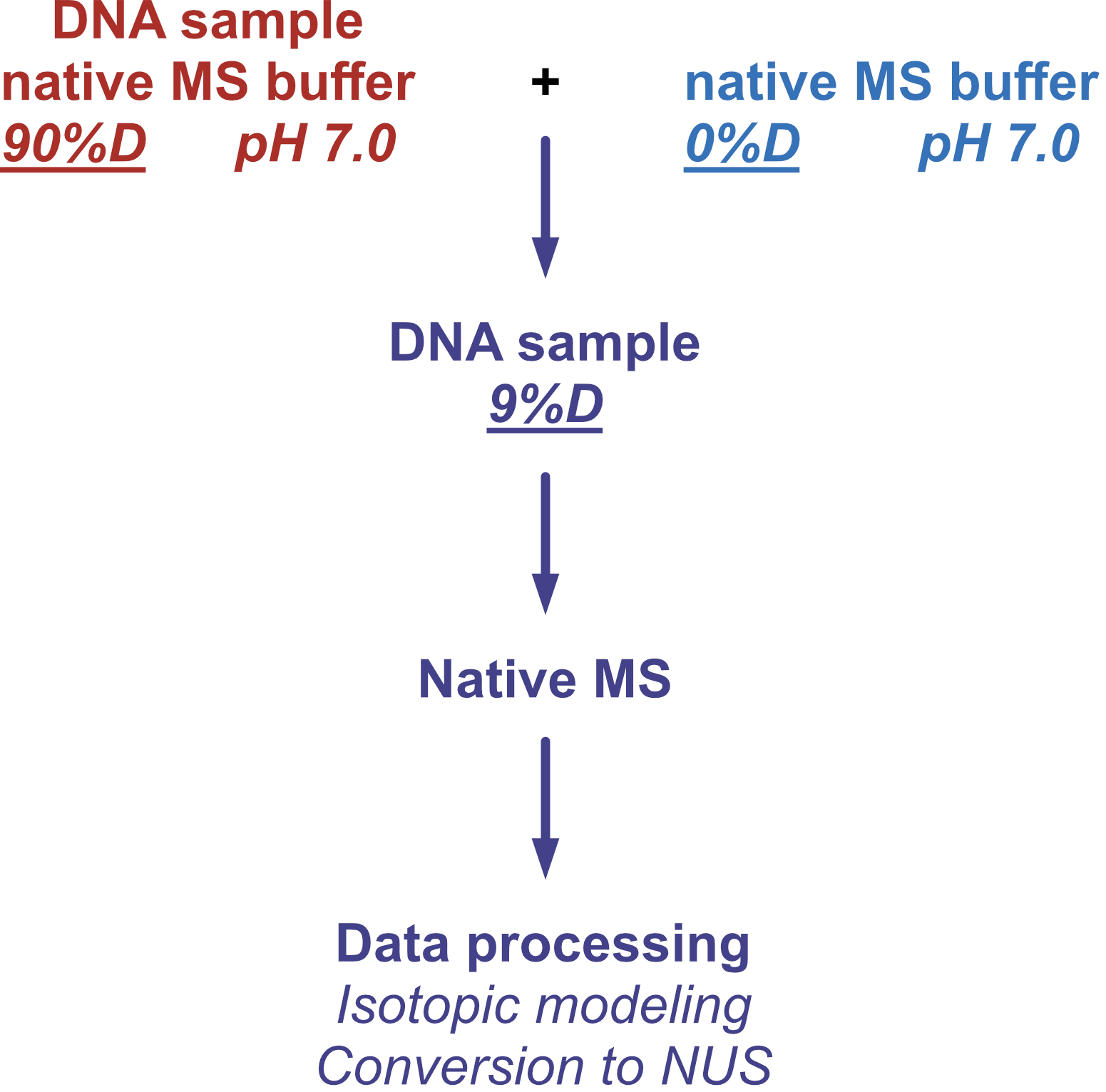
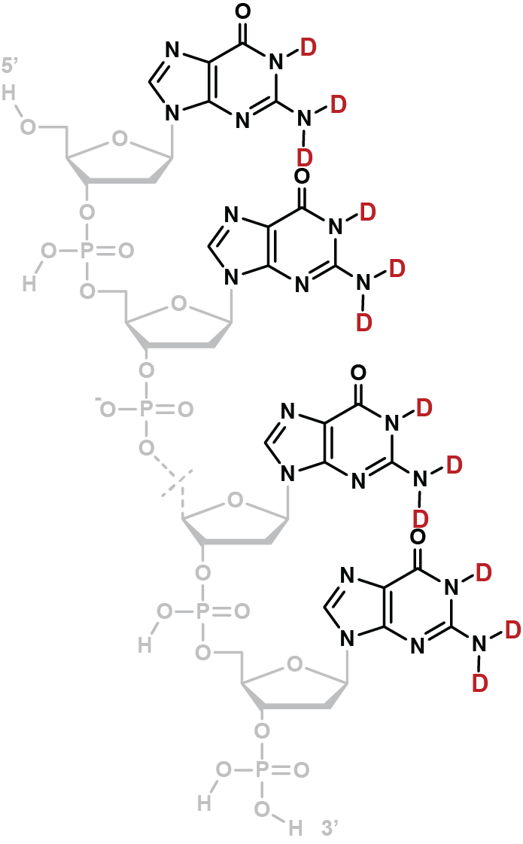
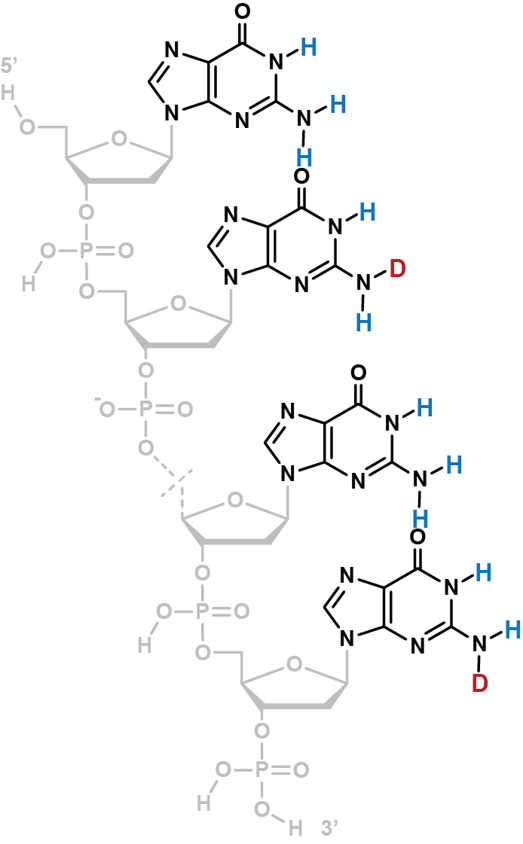
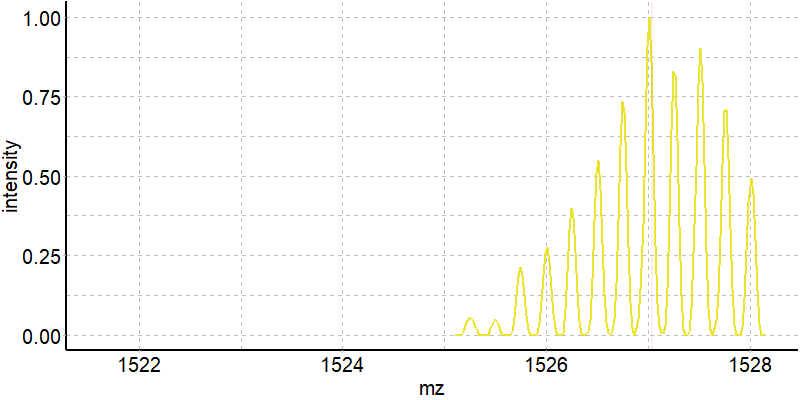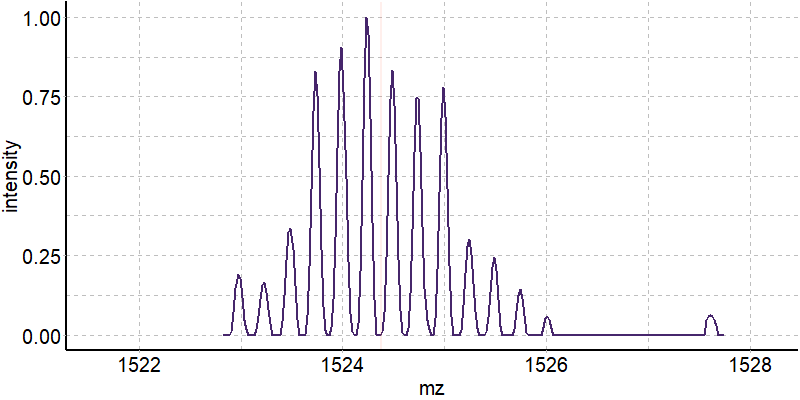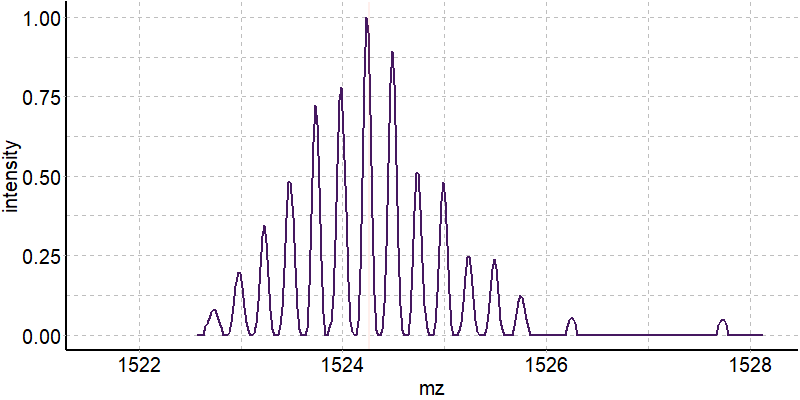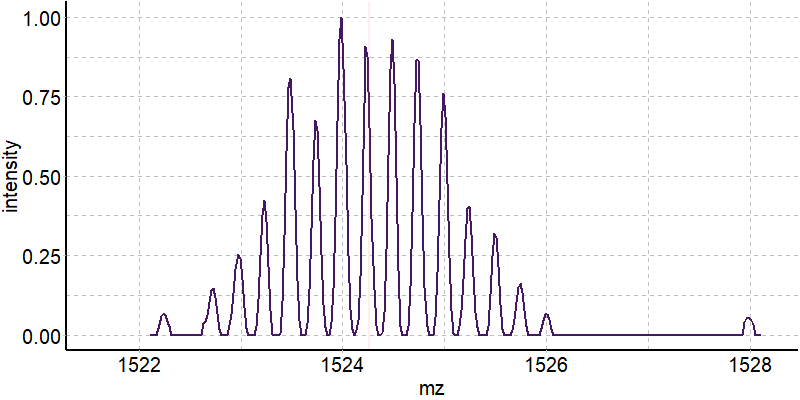
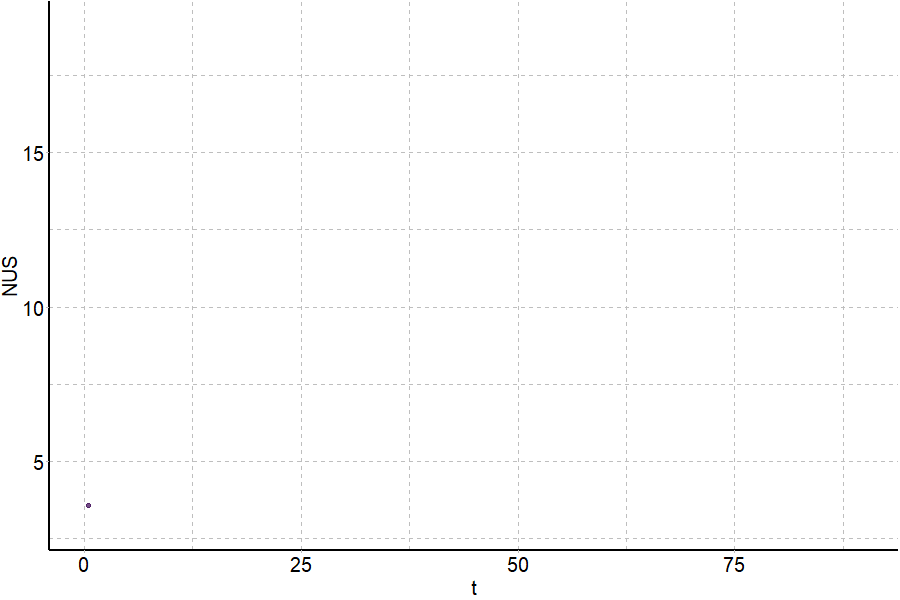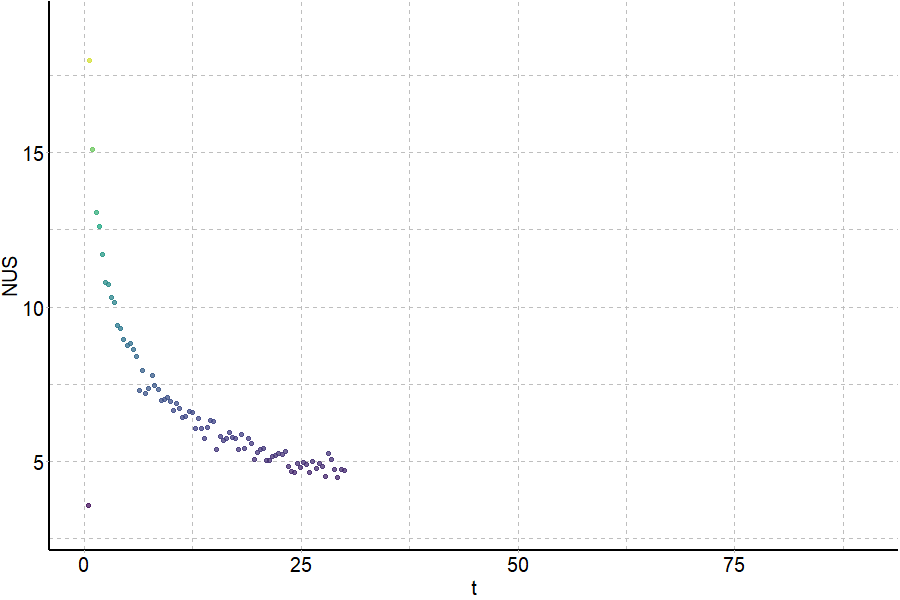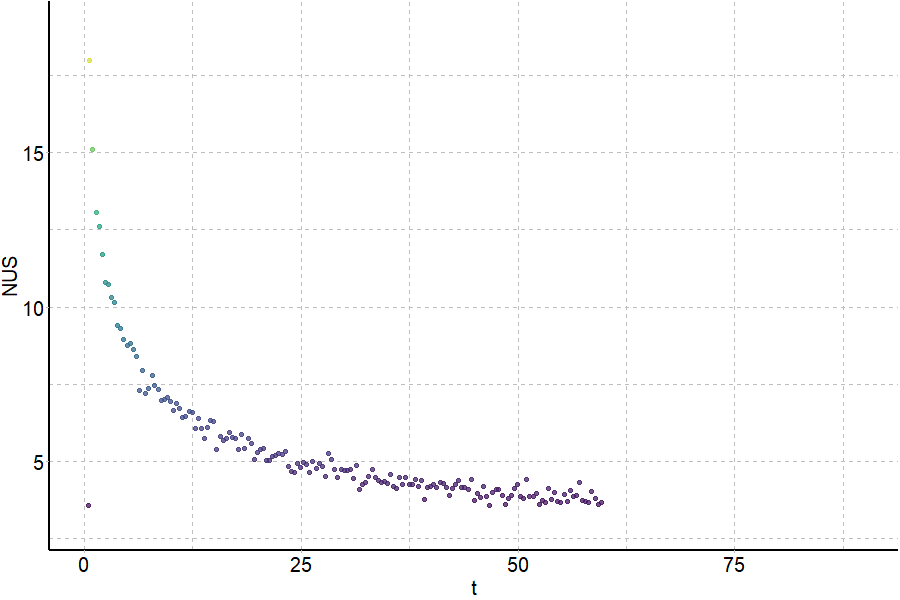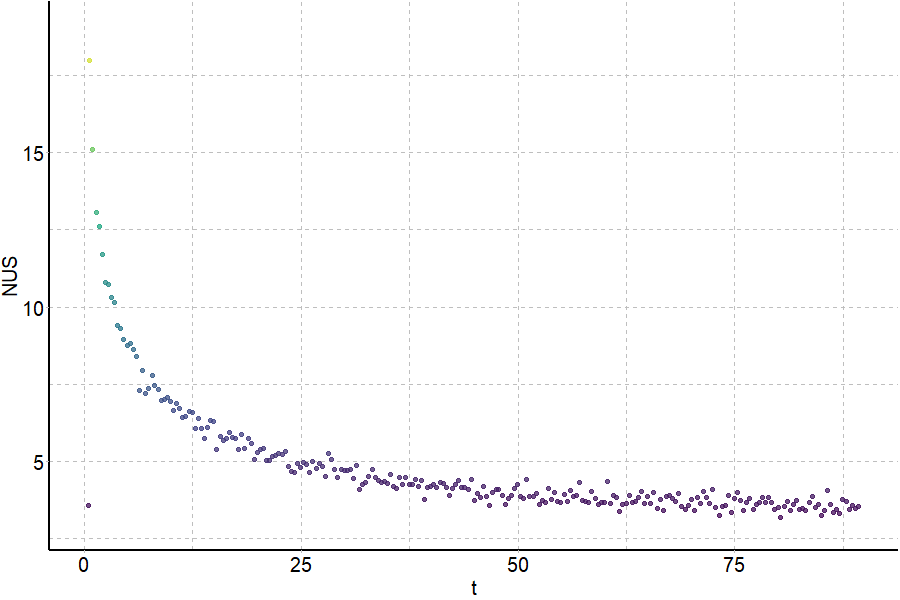
Exchange rates are sensitive to folding
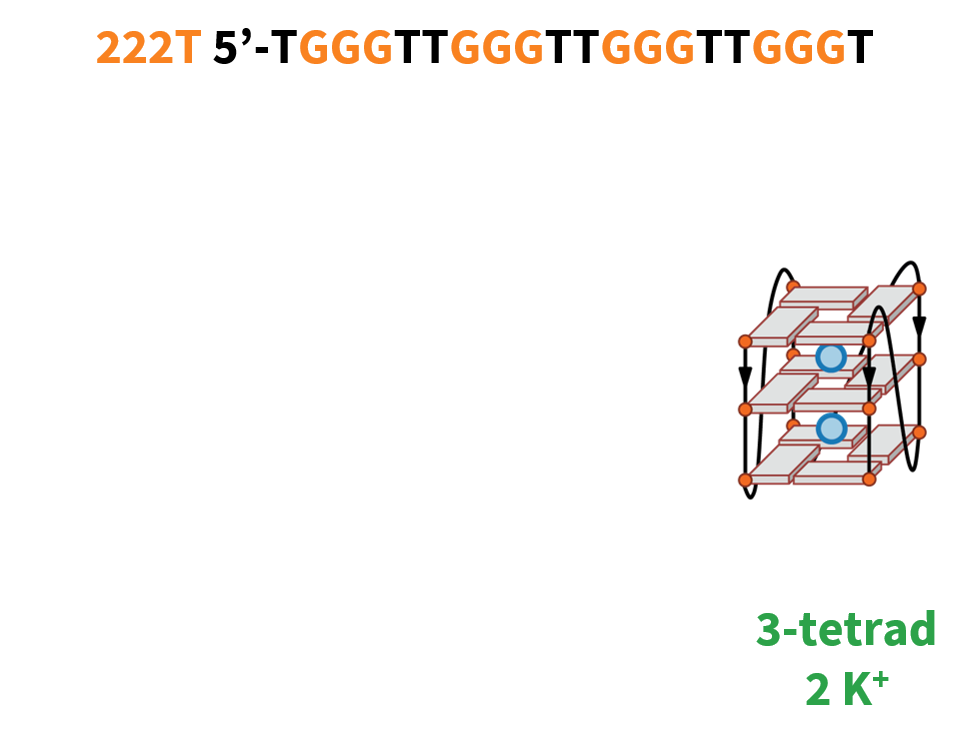
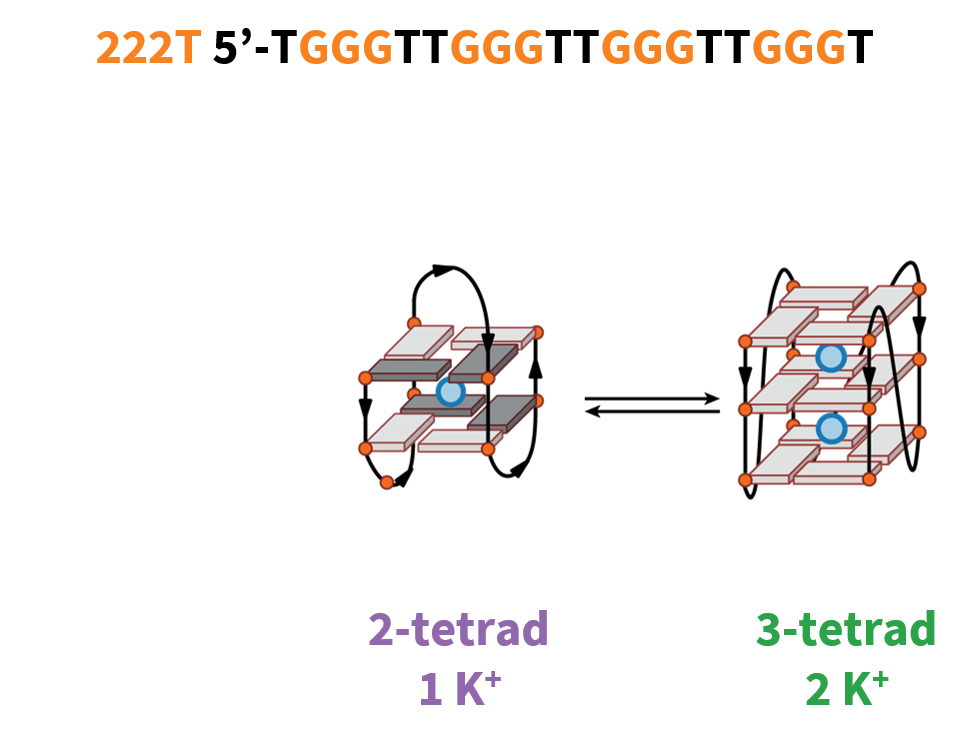
The number of protected sites scales
with the number of tetrads
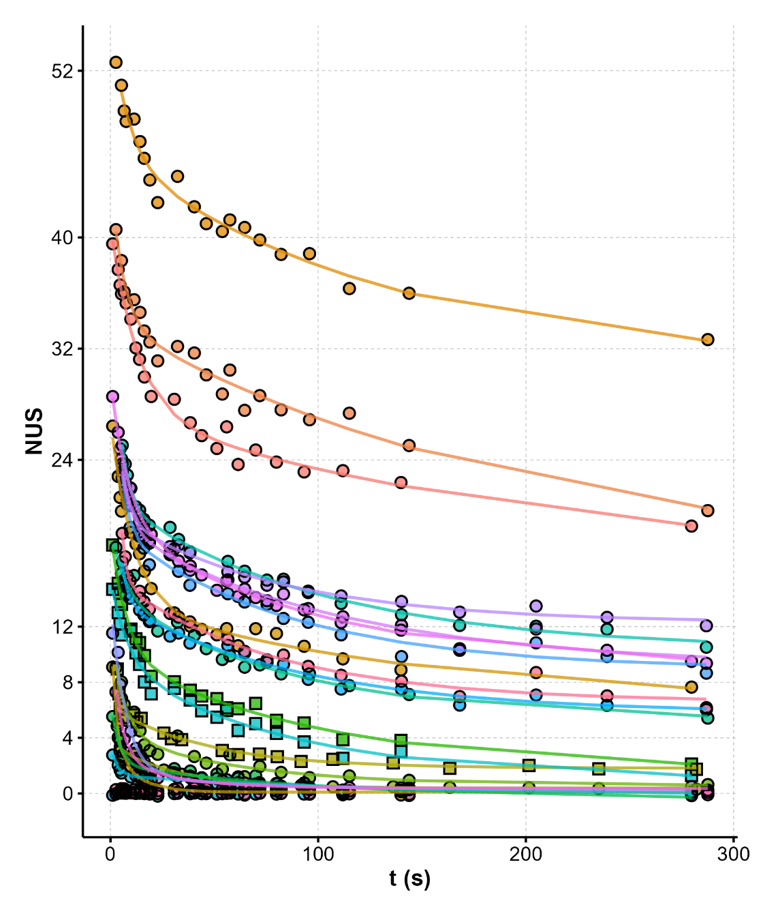
The number of protected sites scales
with the number of tetrads
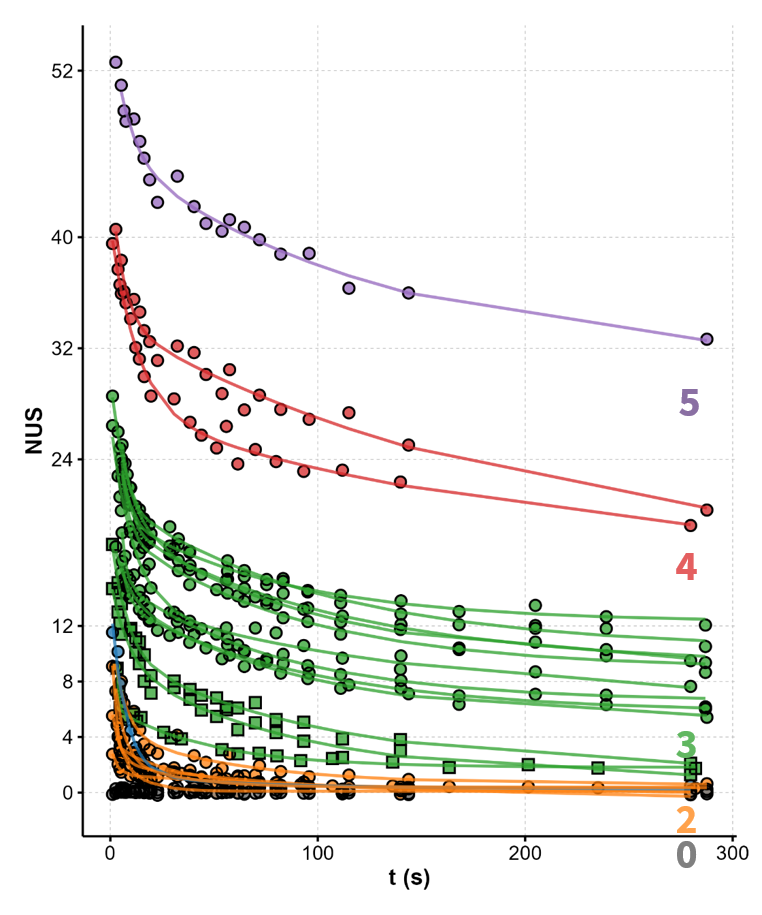
The number of protected sites scales
with the number of tetrads
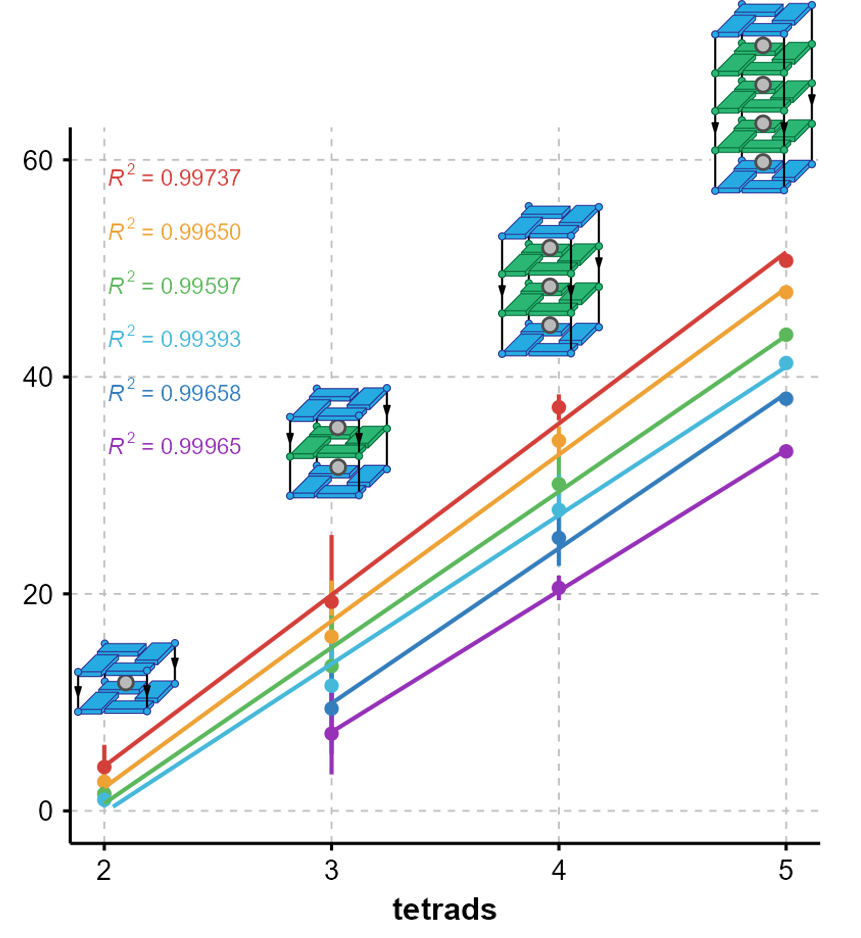
Exchange protection scales with stability
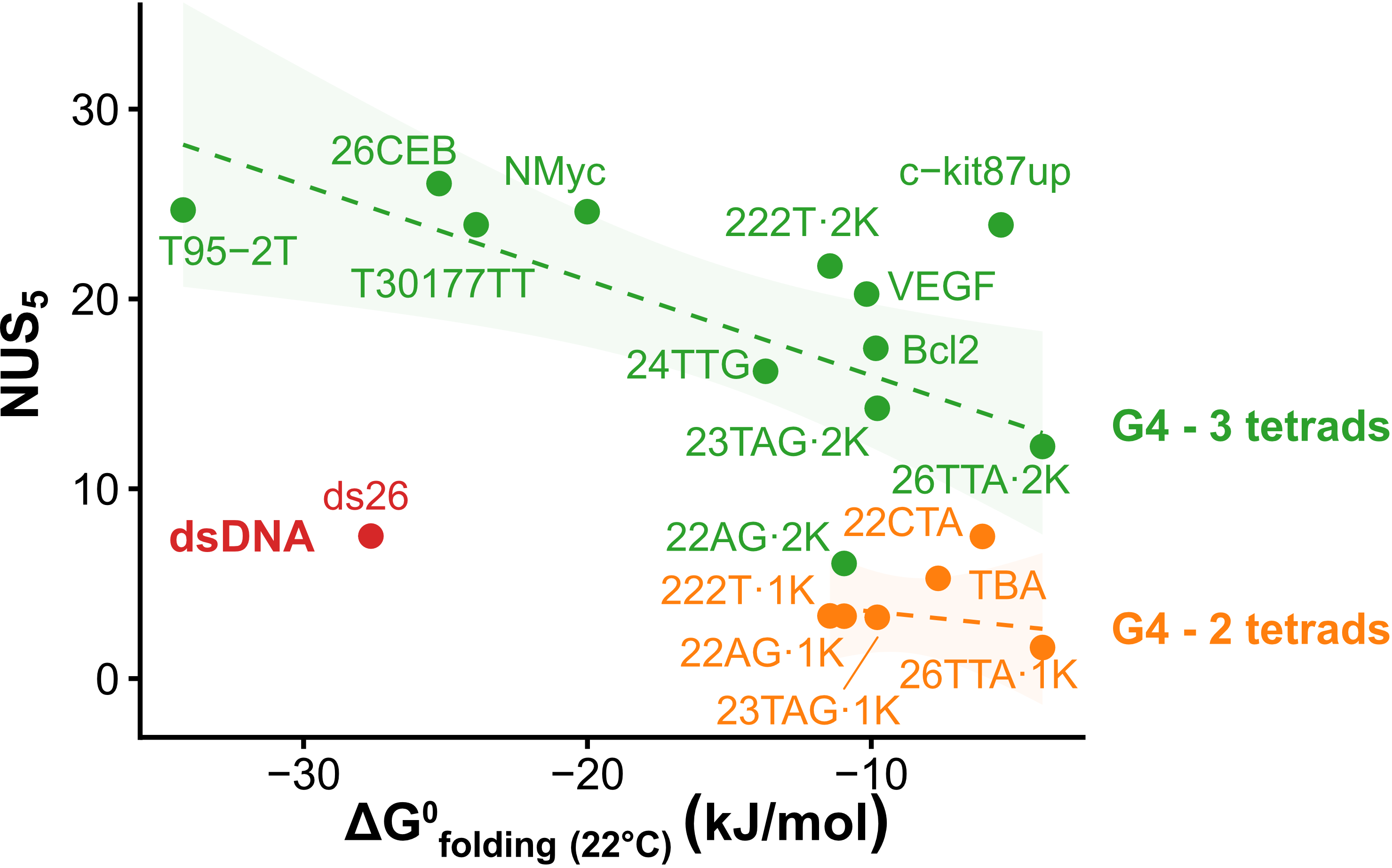
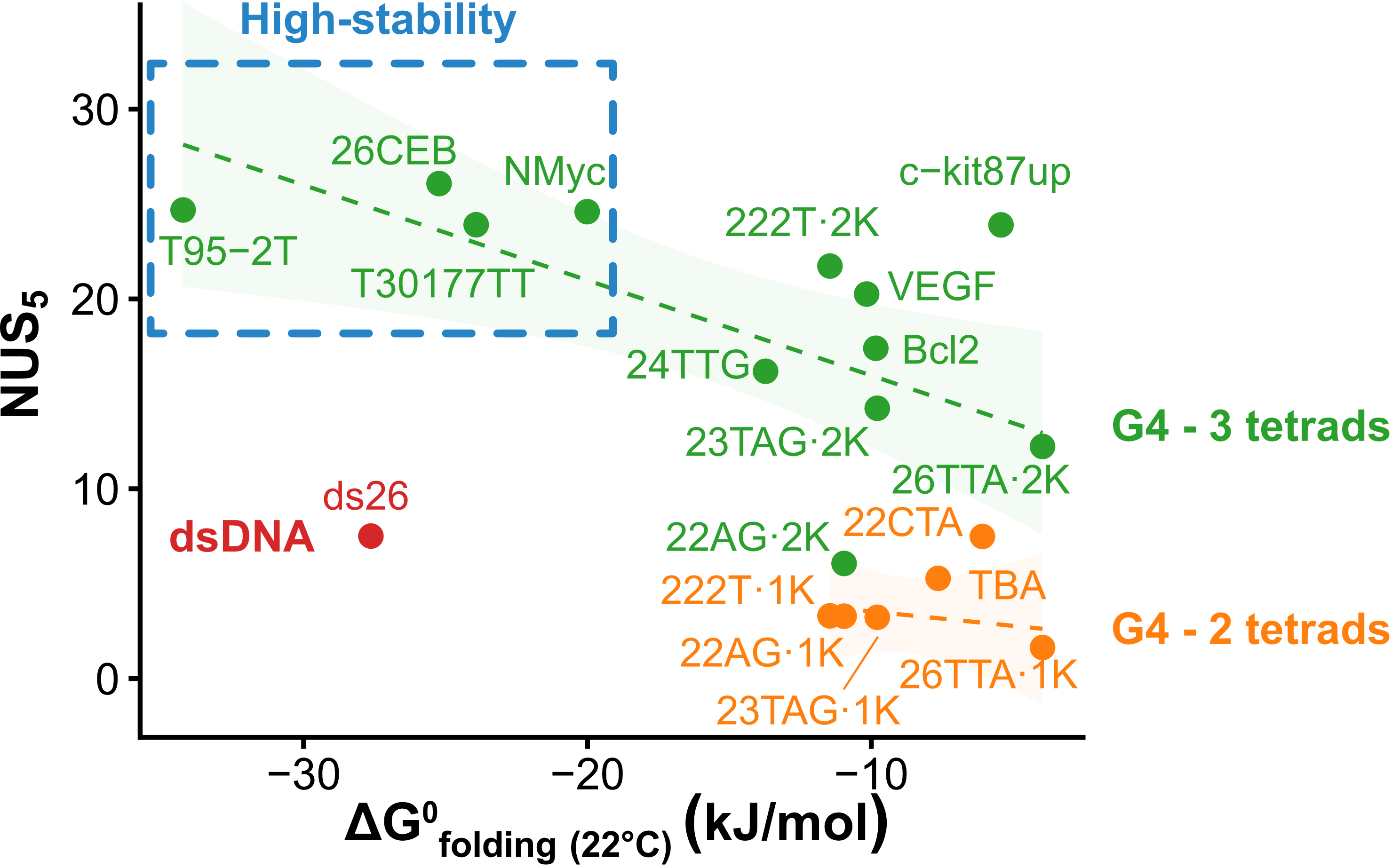
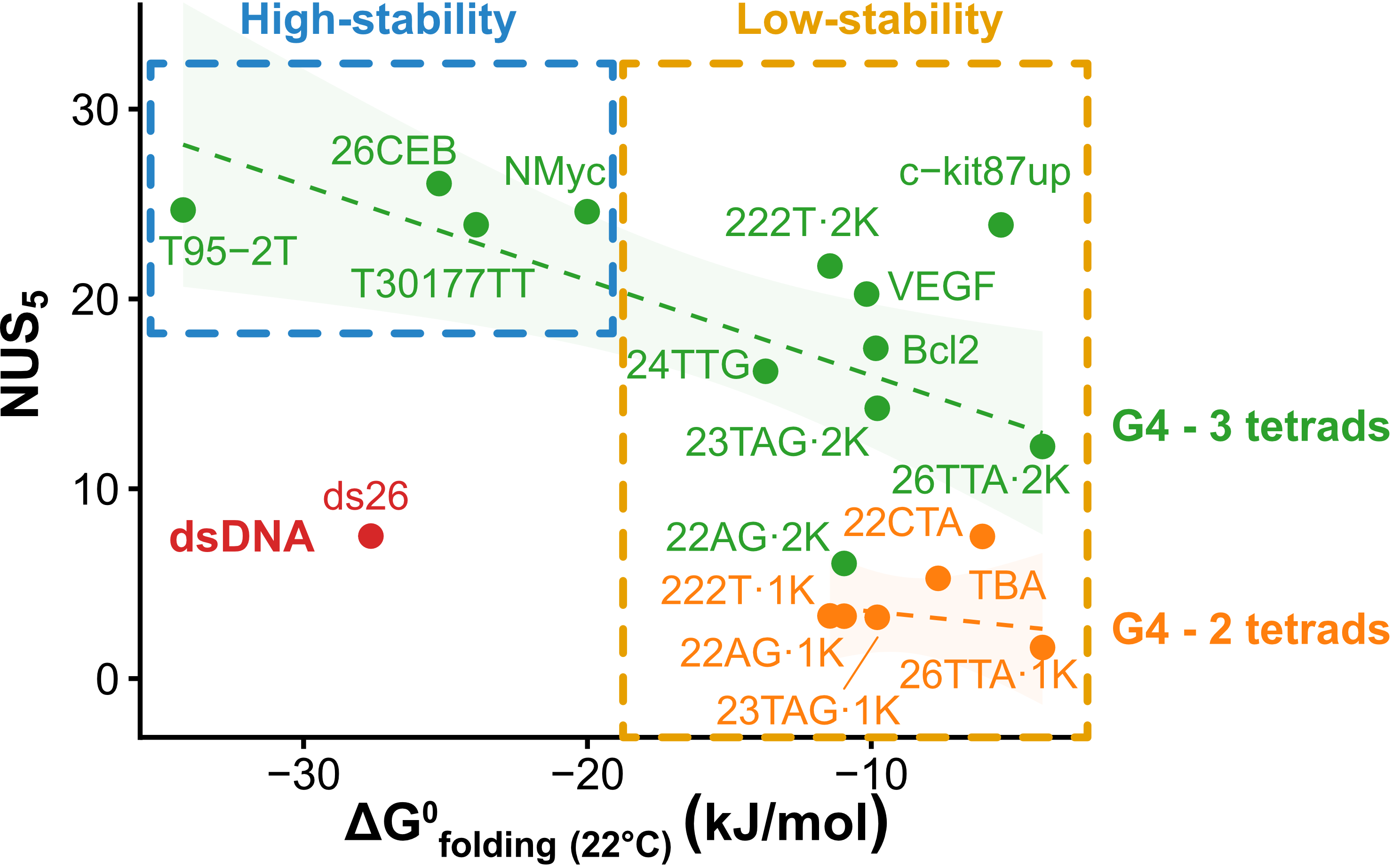
Higher stability G4s exchange via local fluctuations
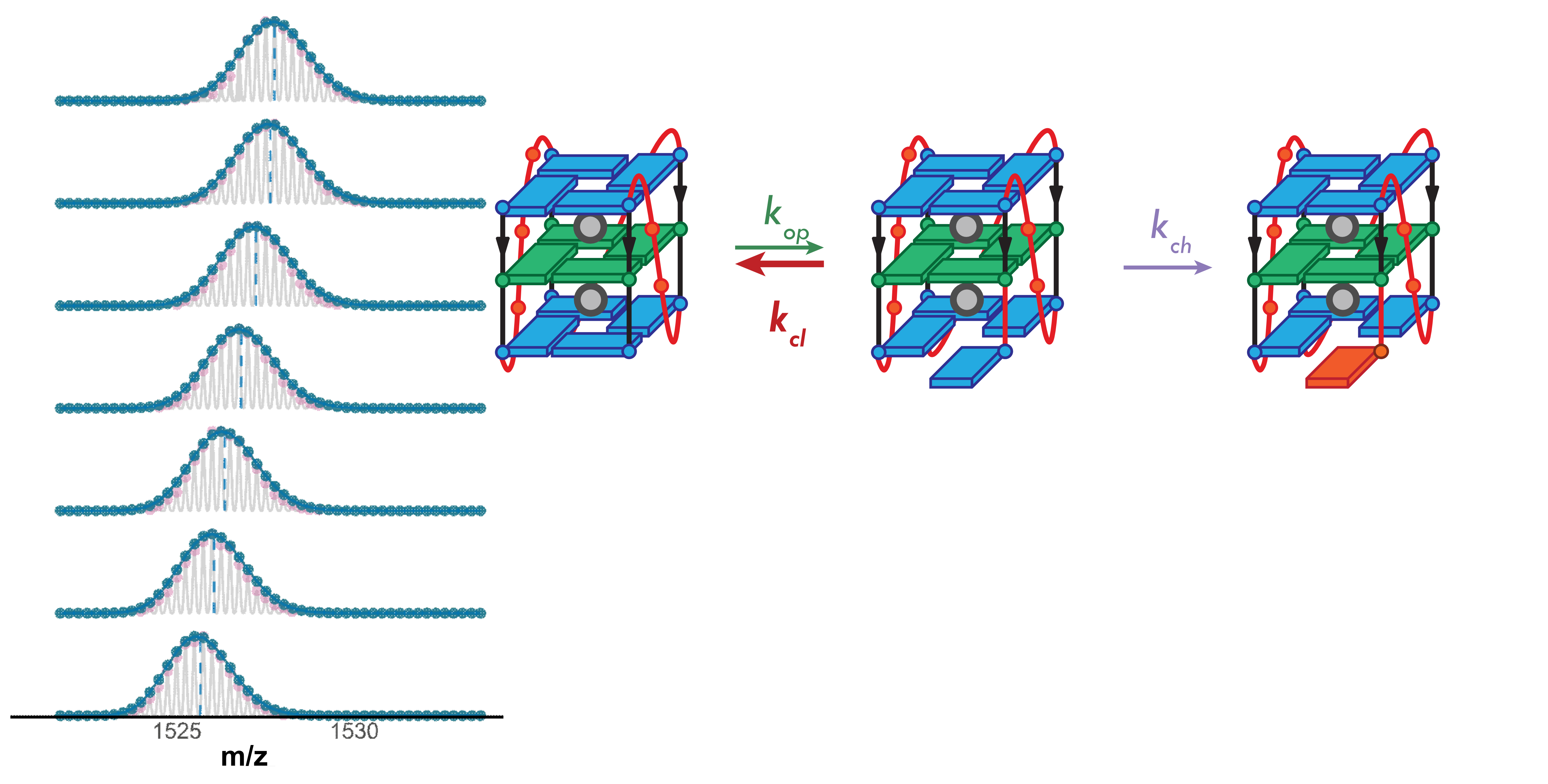
Lower stability G4s exchange via unfolded conformers
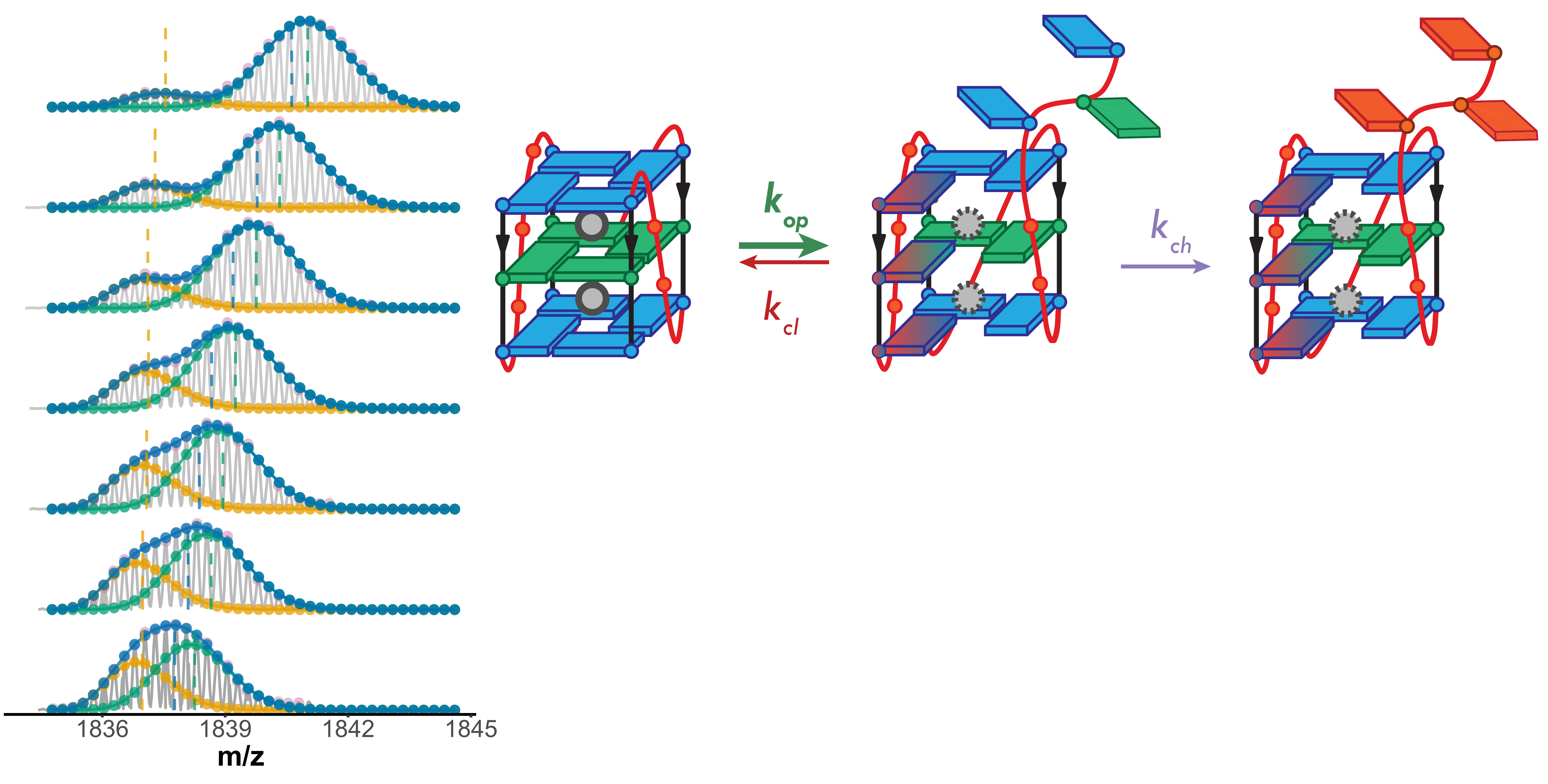
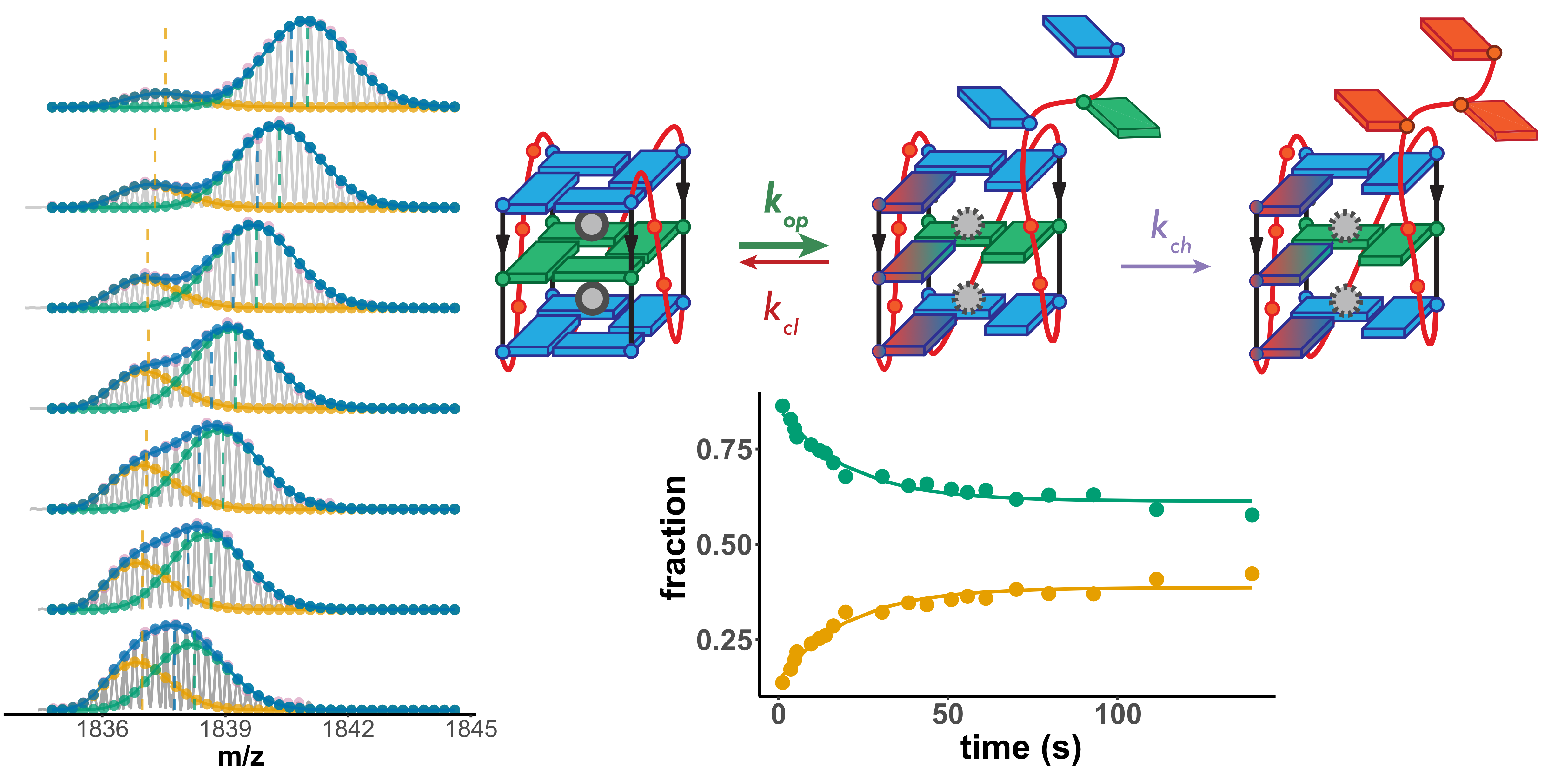
Exchange-competent species
resemble folding intermediates

Drift-tube IMS can separate analytes by shape

Drift-tube IMS can separate analytes by shape
Minor conformers revealed by HDX/IMS
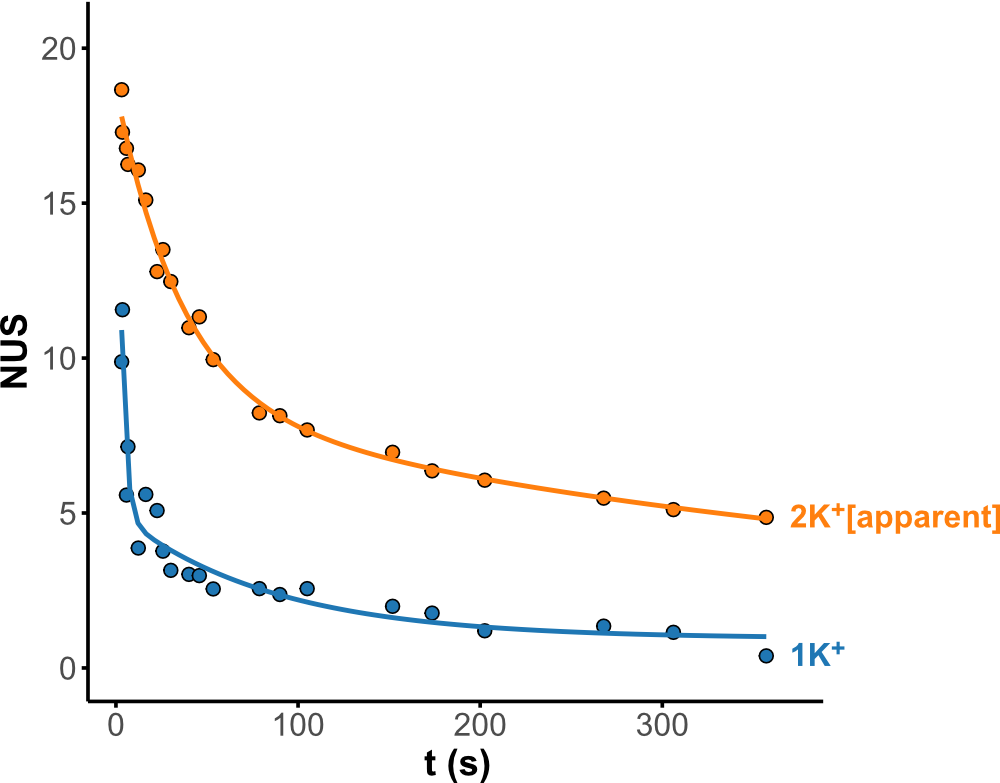
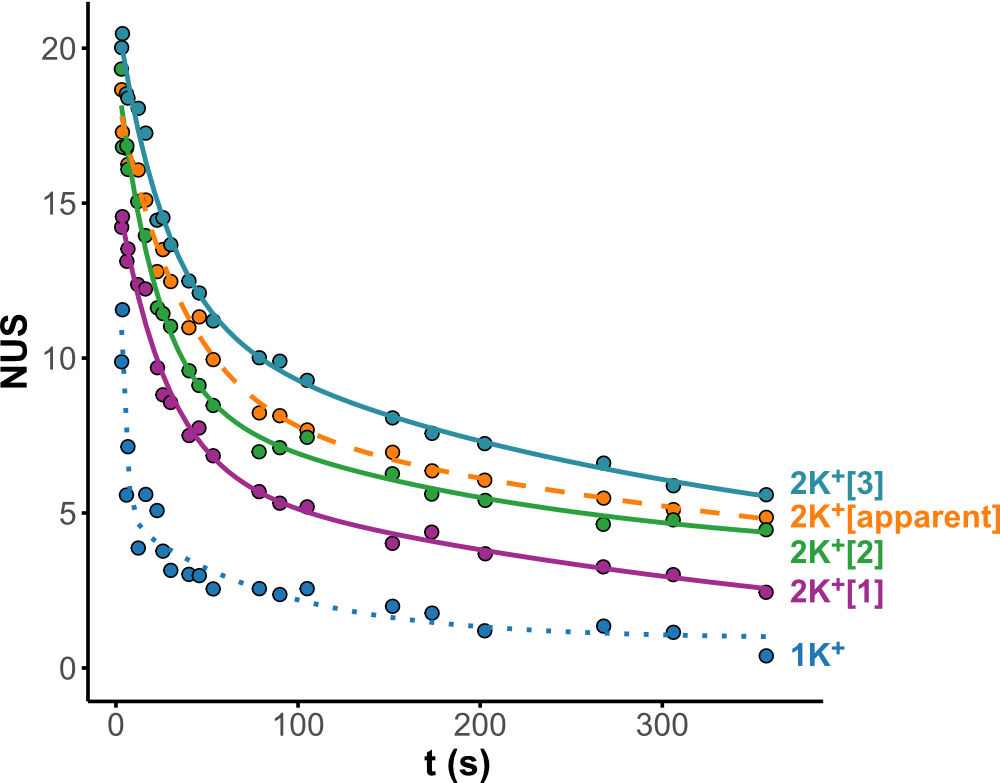
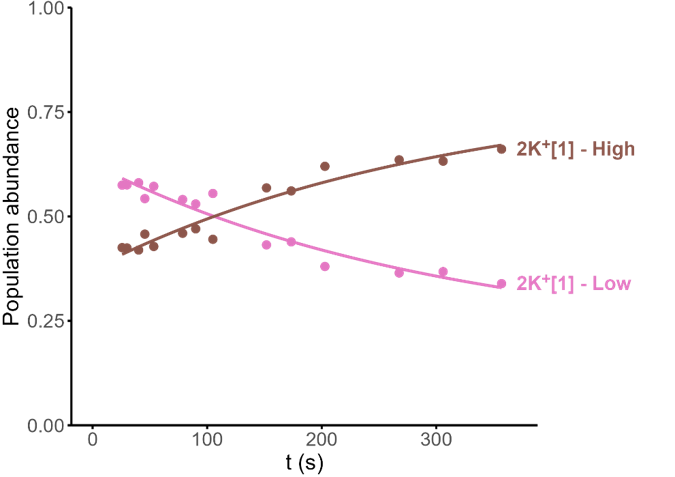
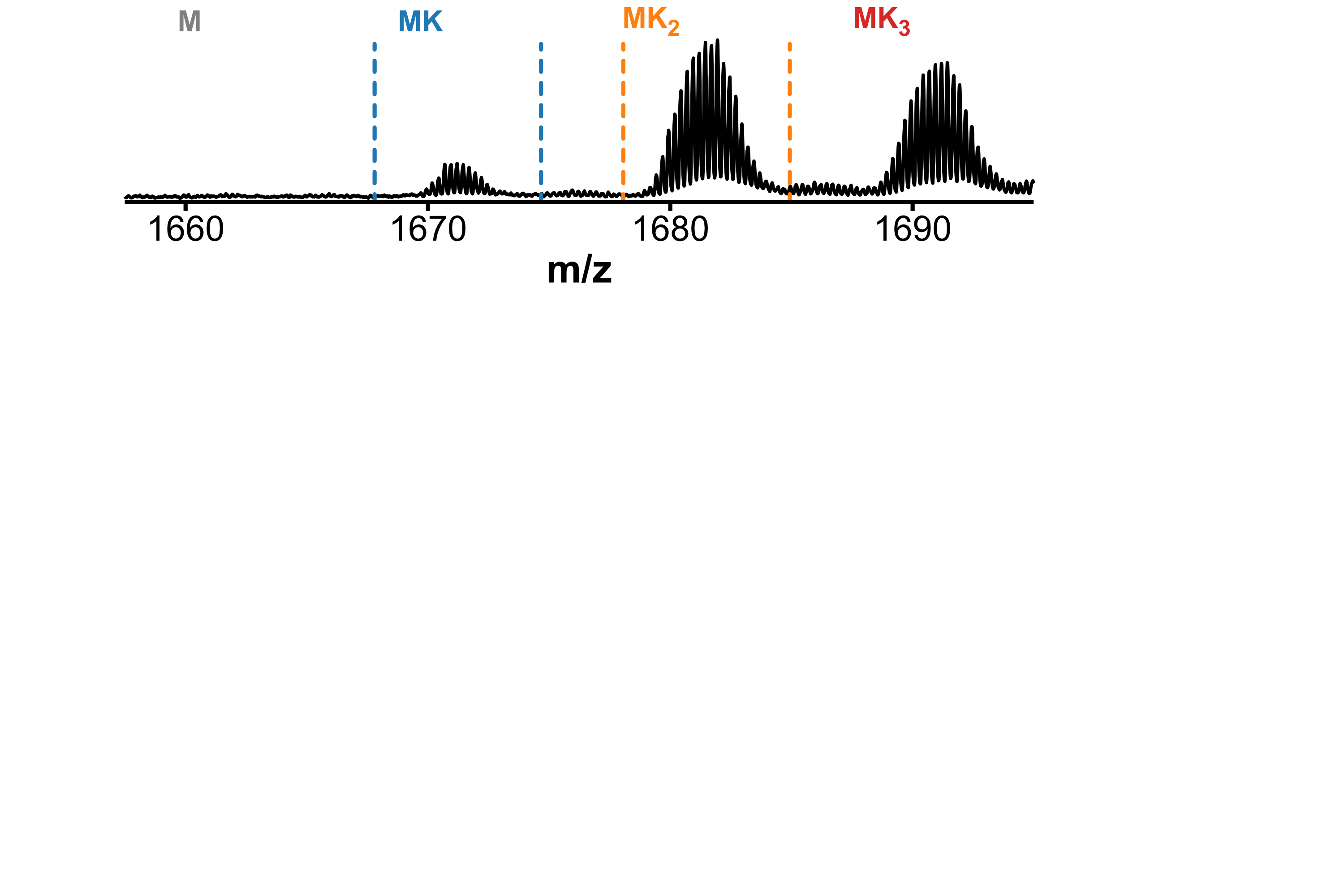
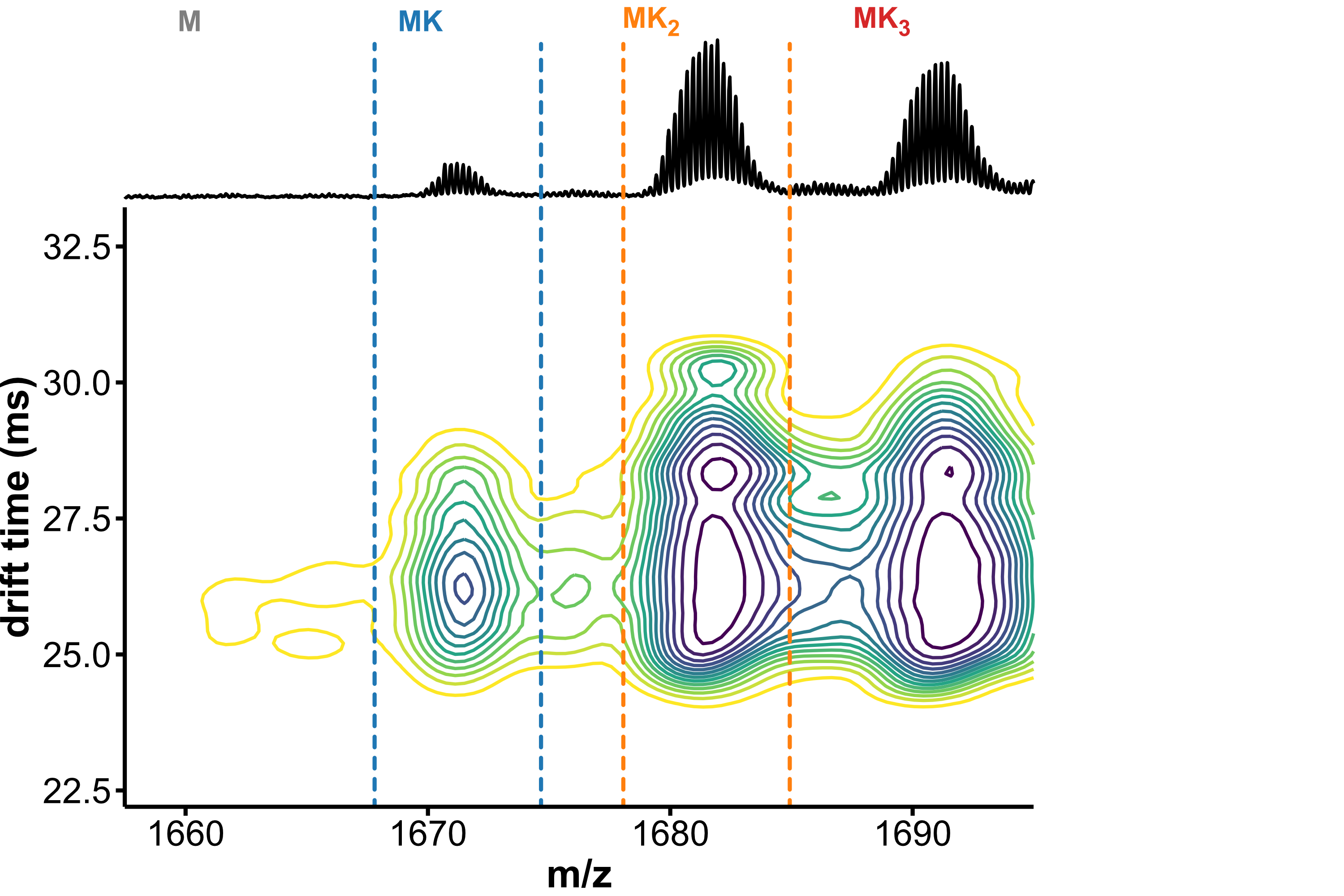
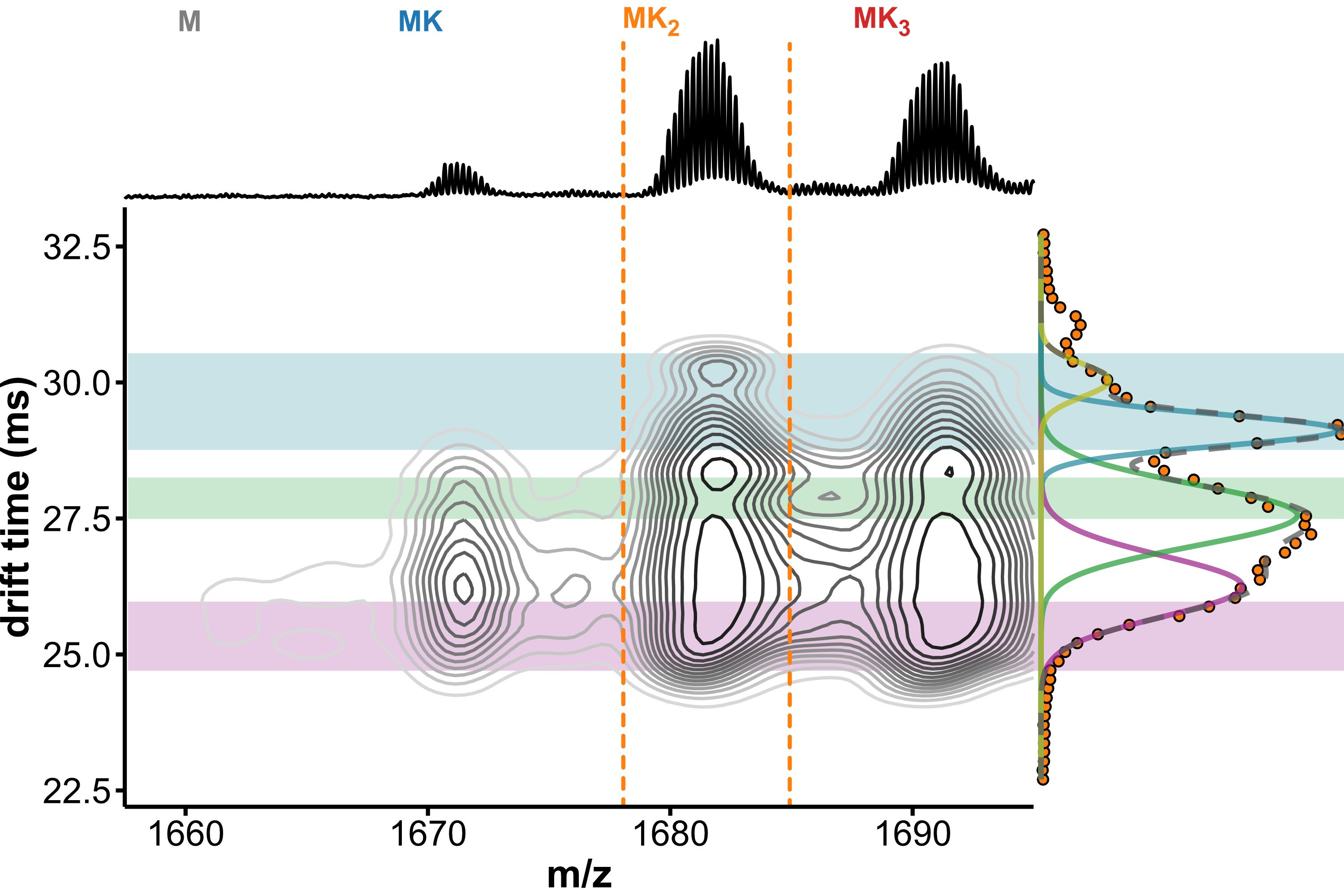
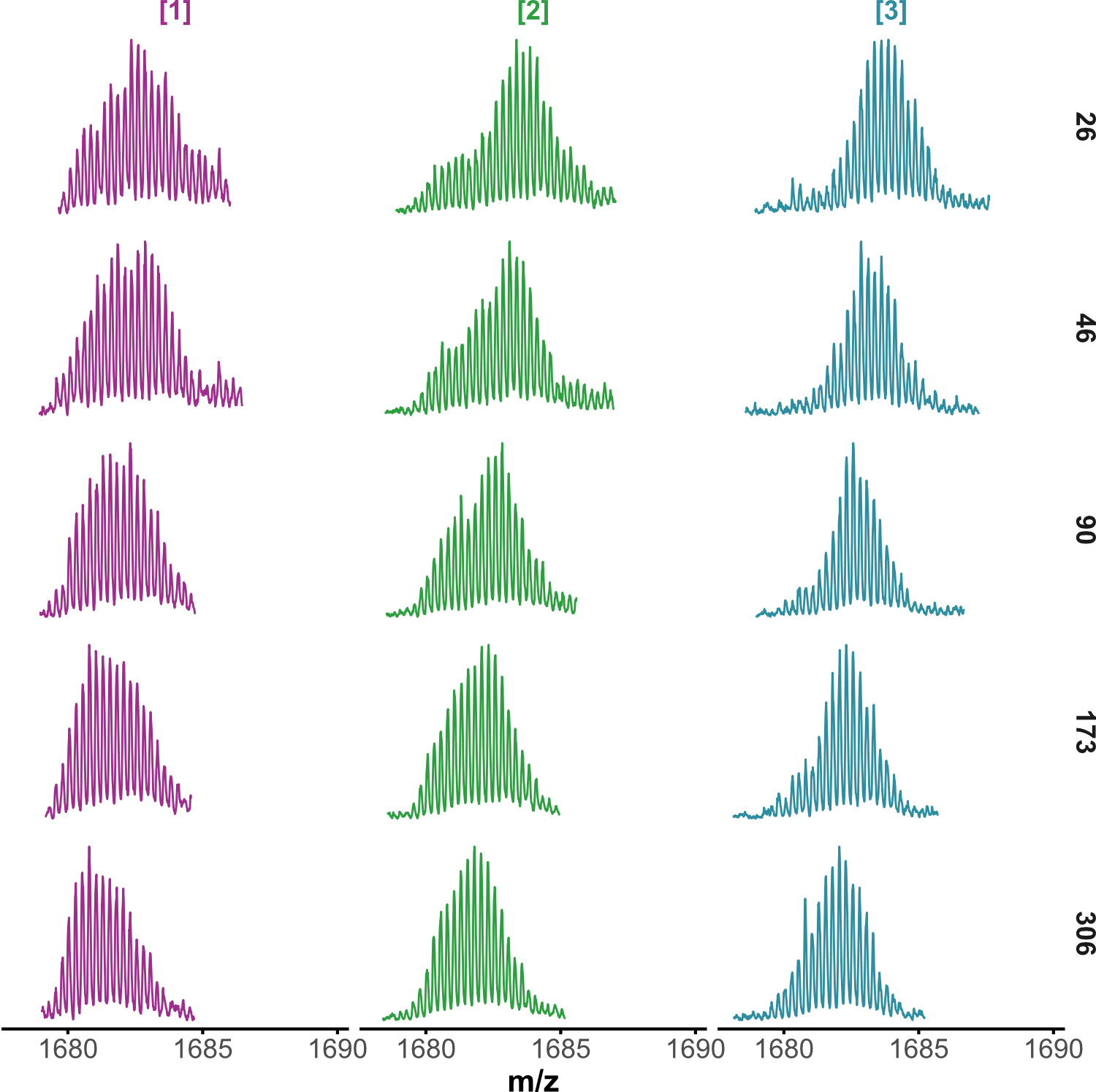
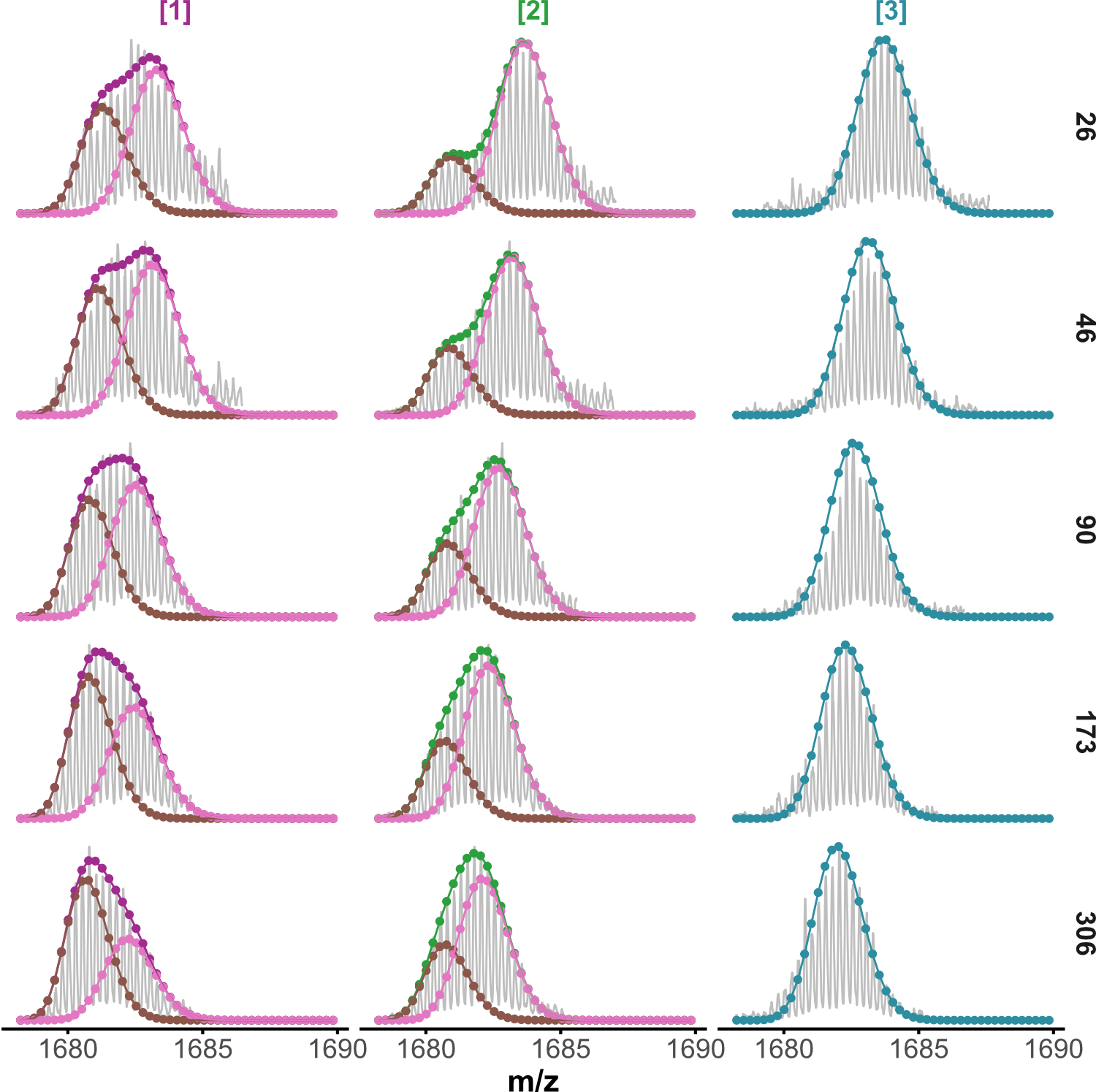


HDX kinetics inform on binding modes
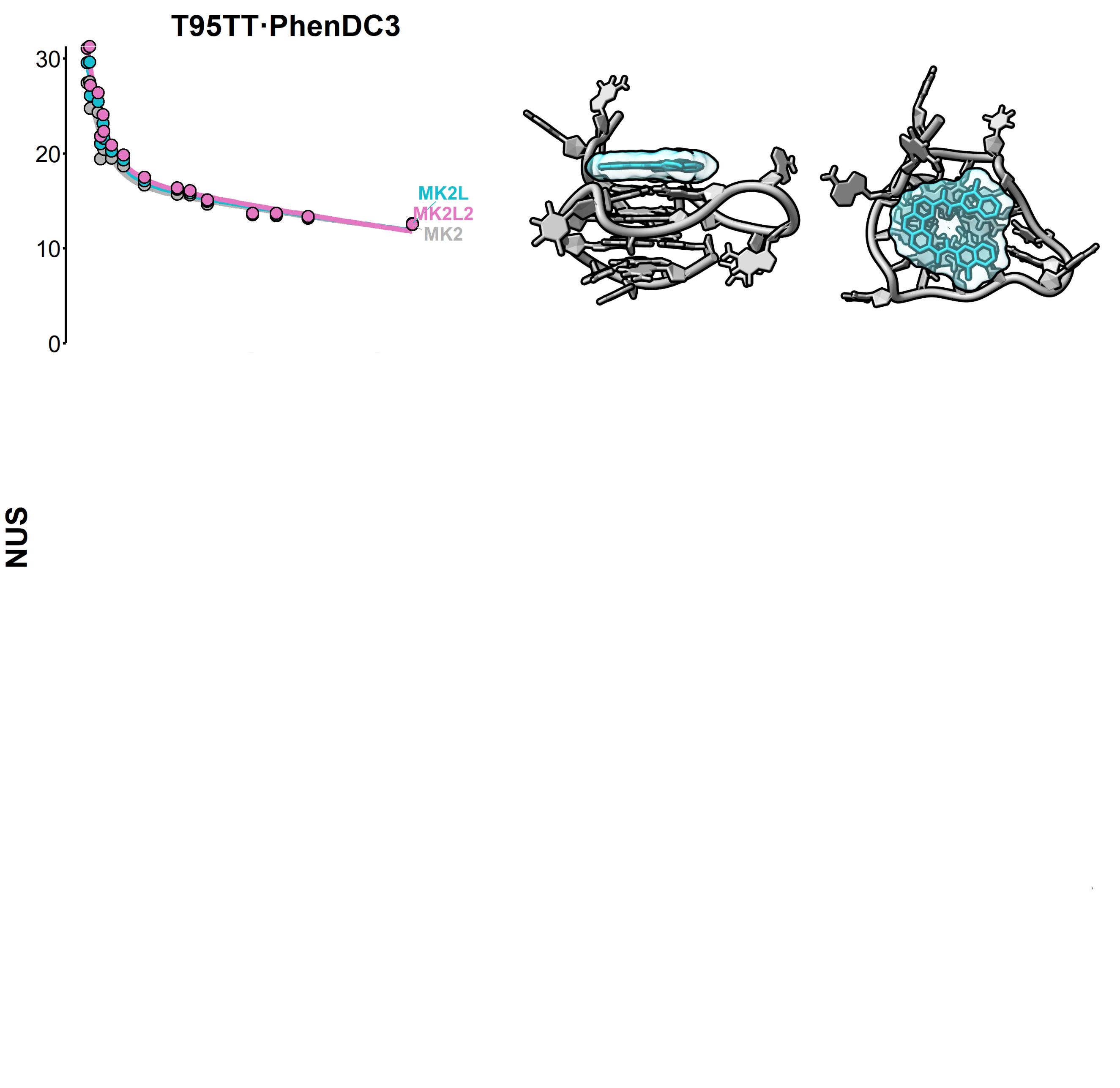
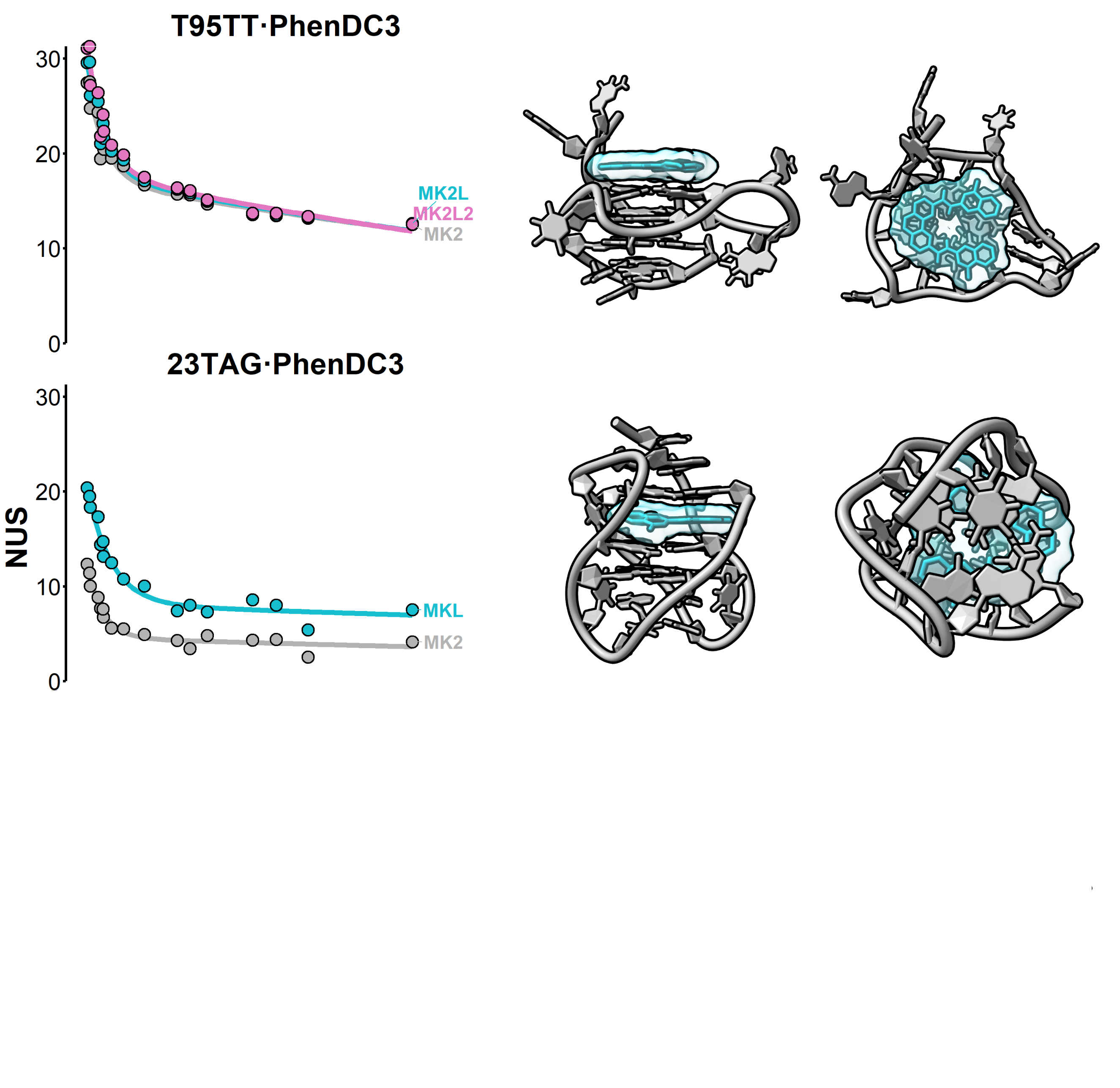
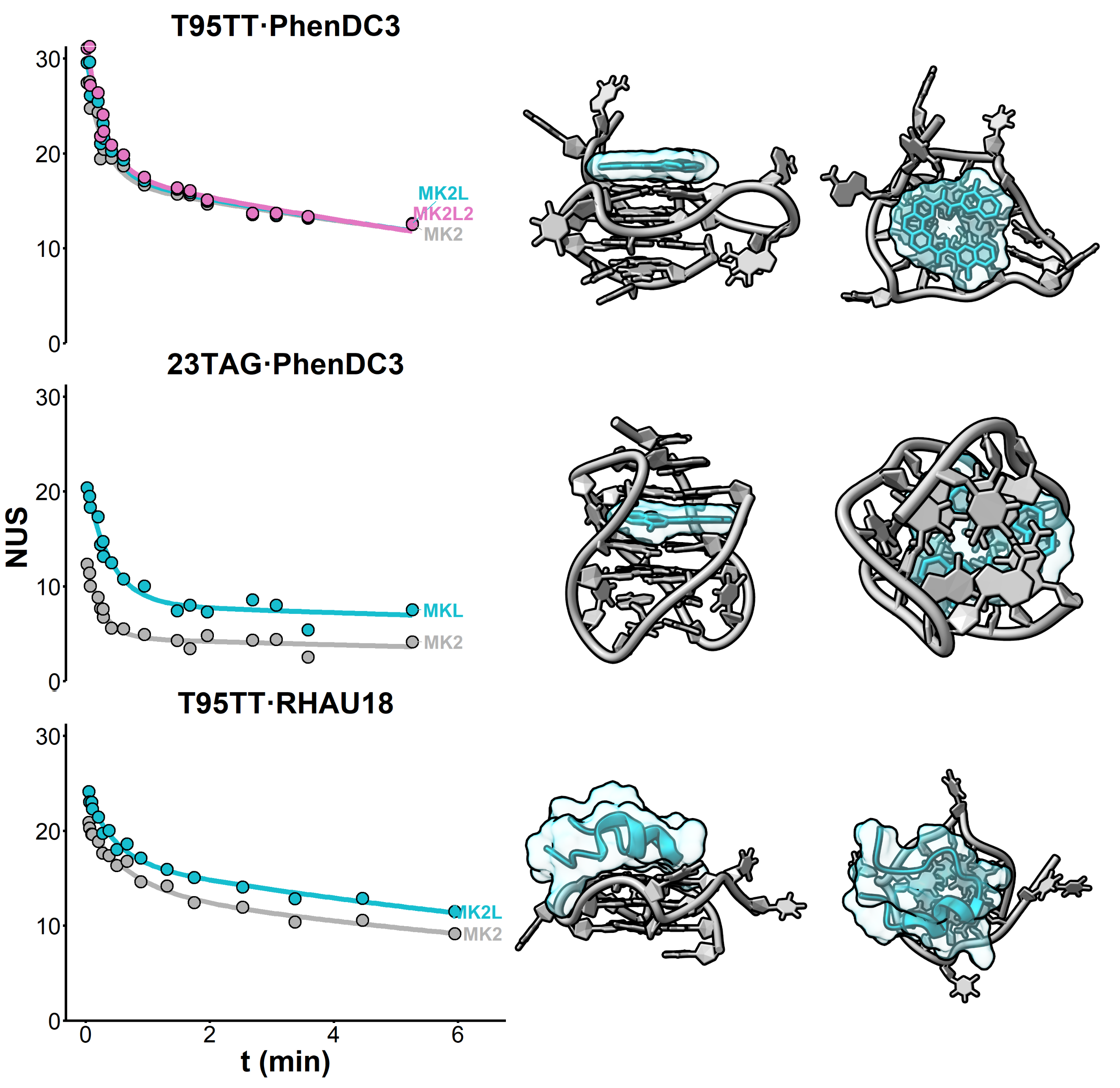

Aptamer structures are seldom characterized
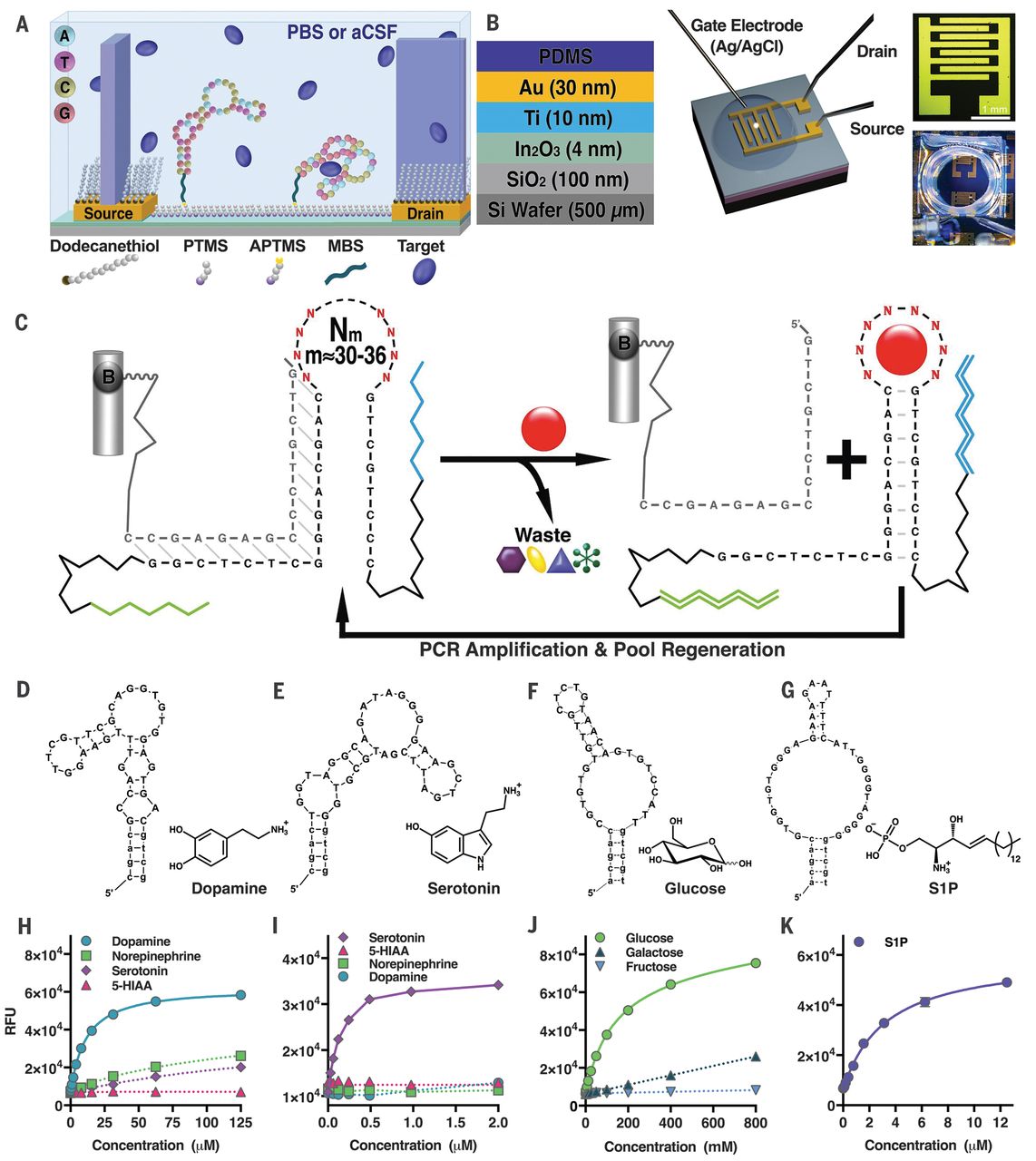
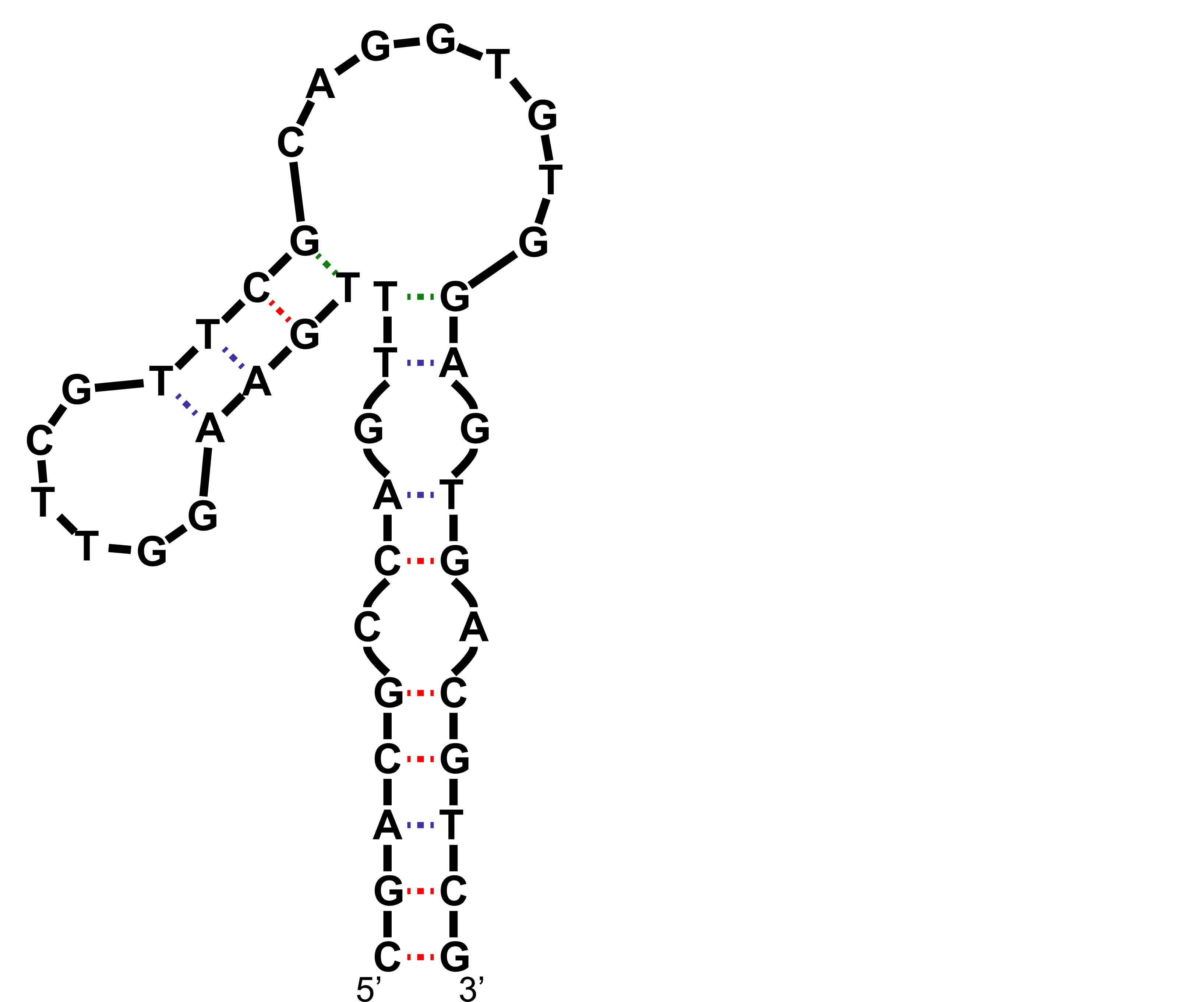
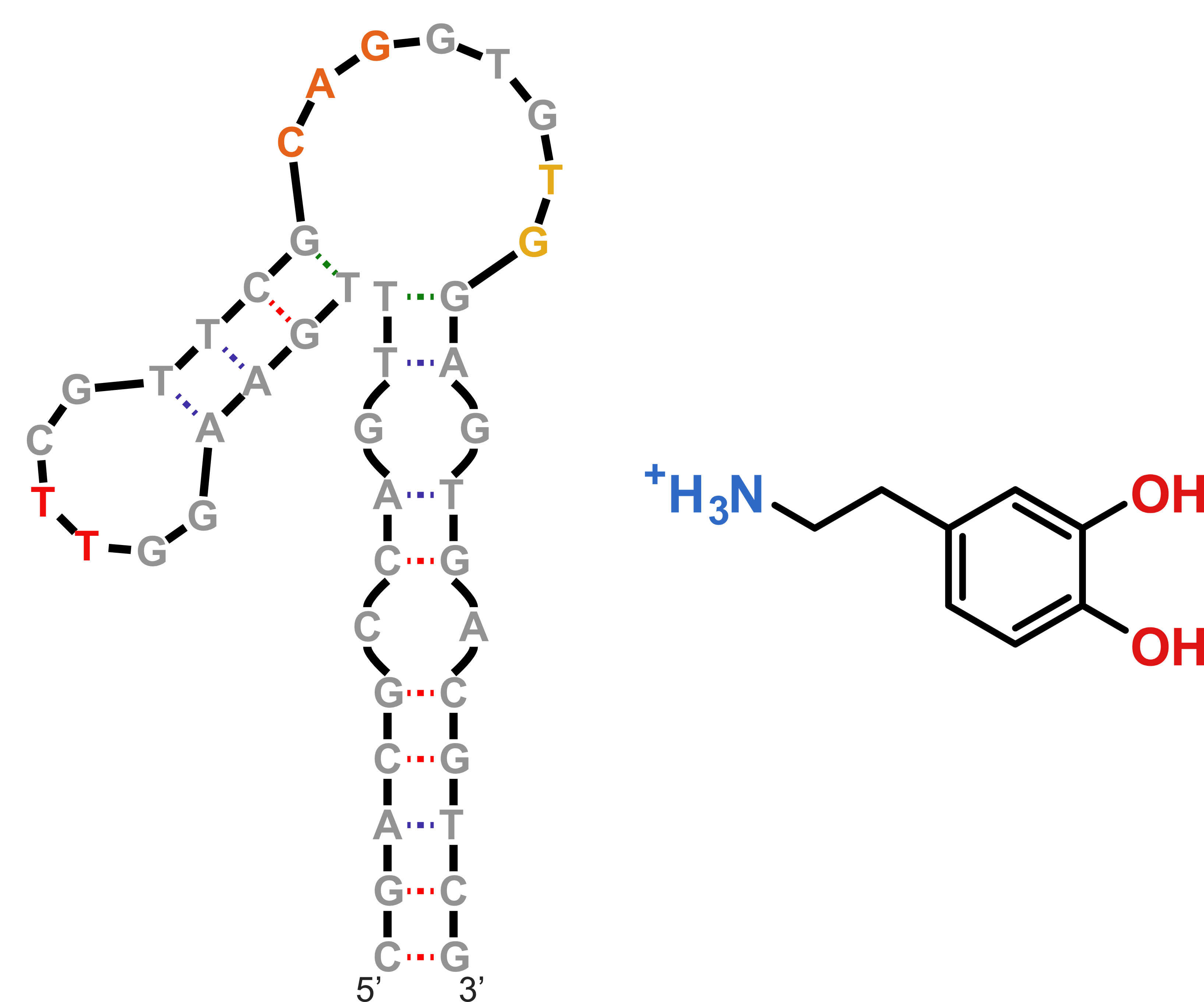
NMR provides (non-canonical !)
starting models
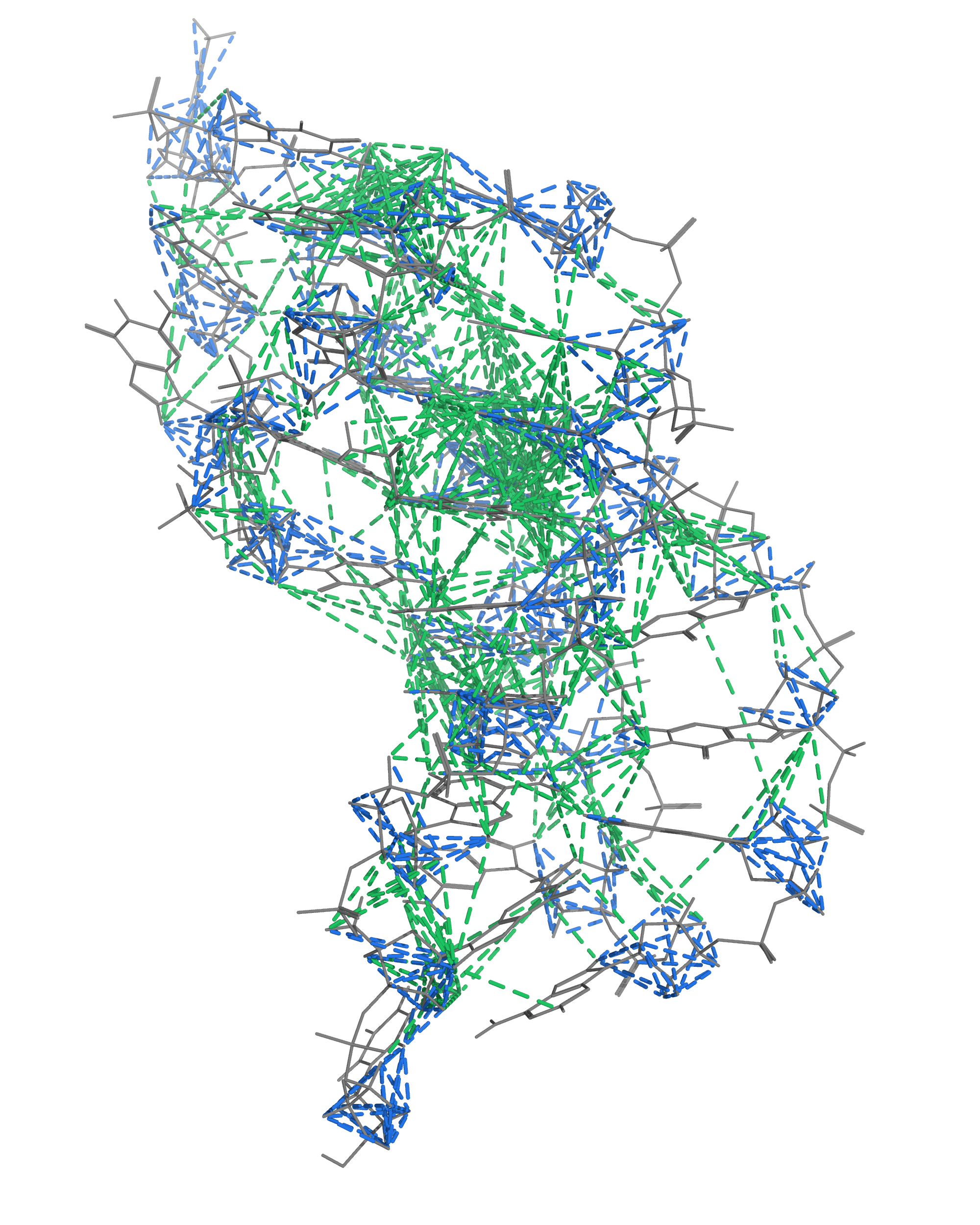
NMR provides many constraints
- DNA (806)
- intra-residue
- inter-residue
- Dopamine (67)
- intramolecular
- intermolecular
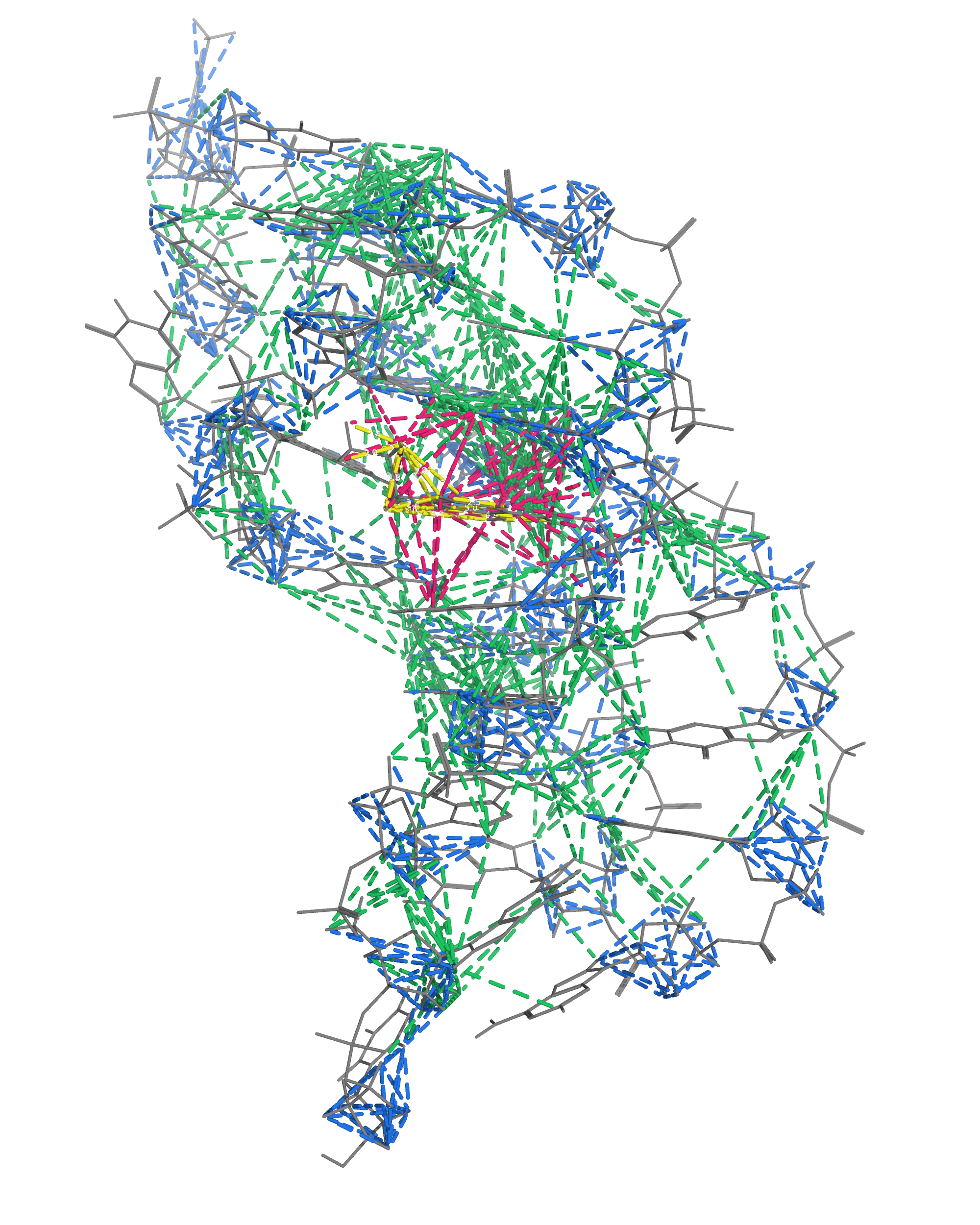

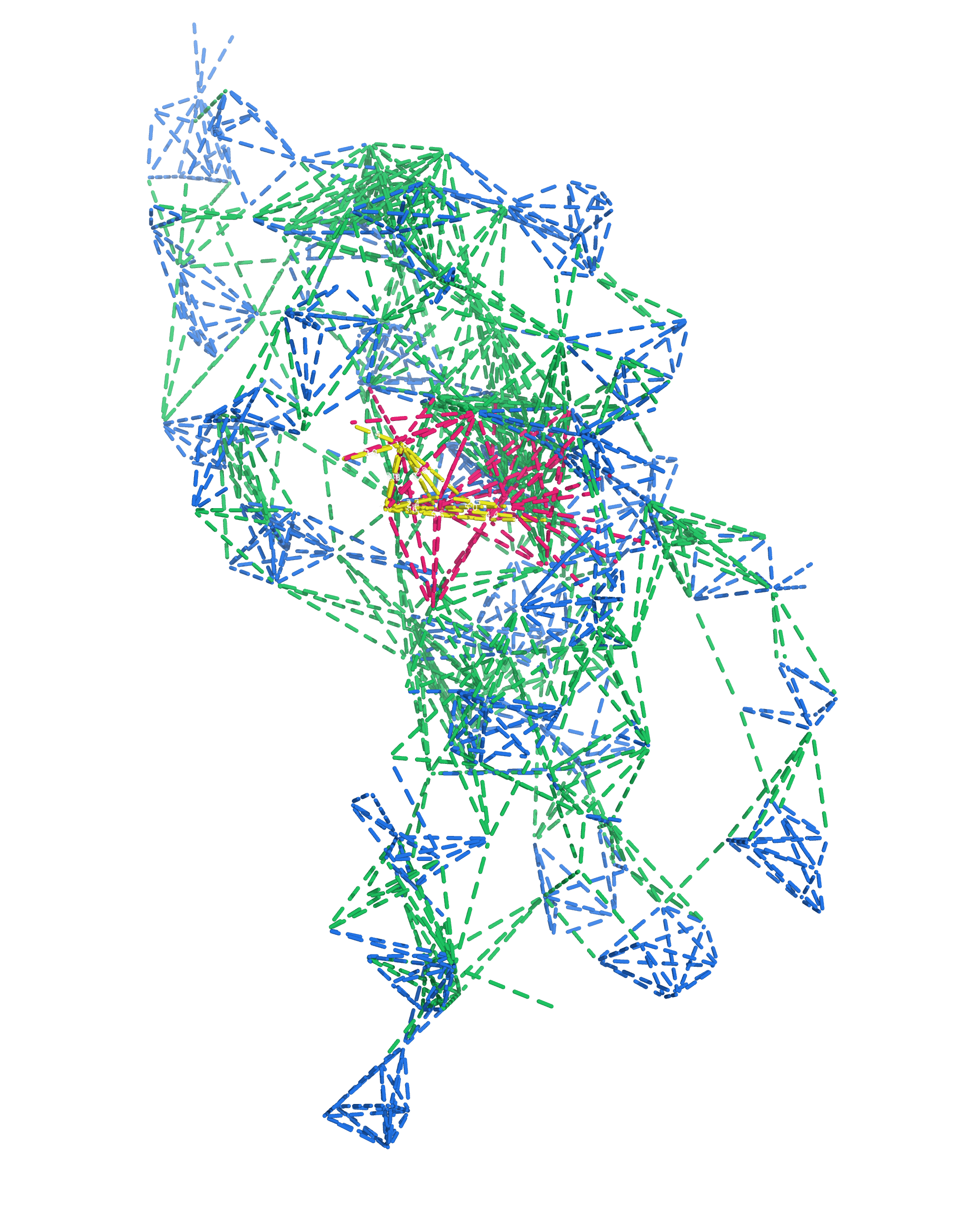
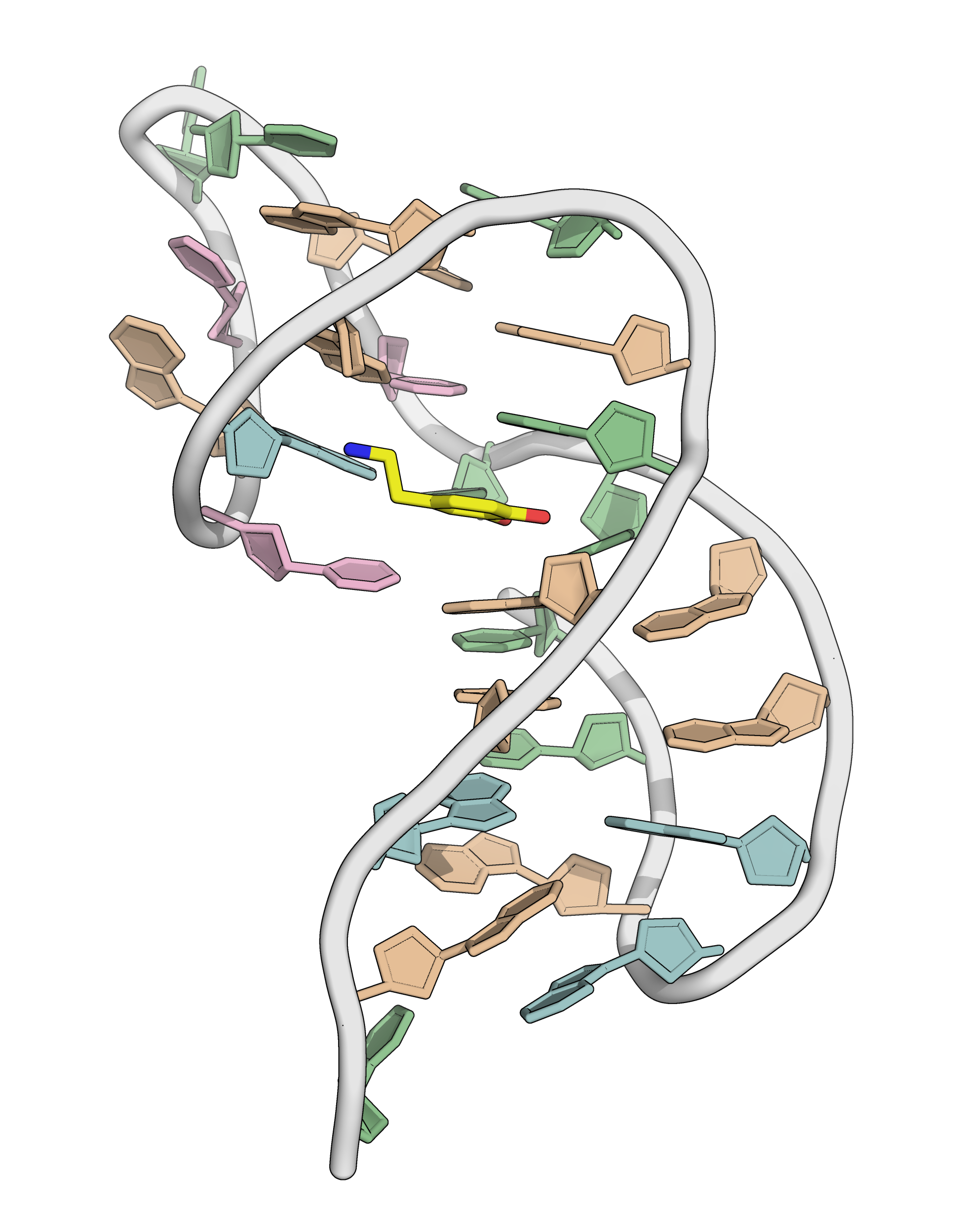
A minimized ensemble that cannot be predicted
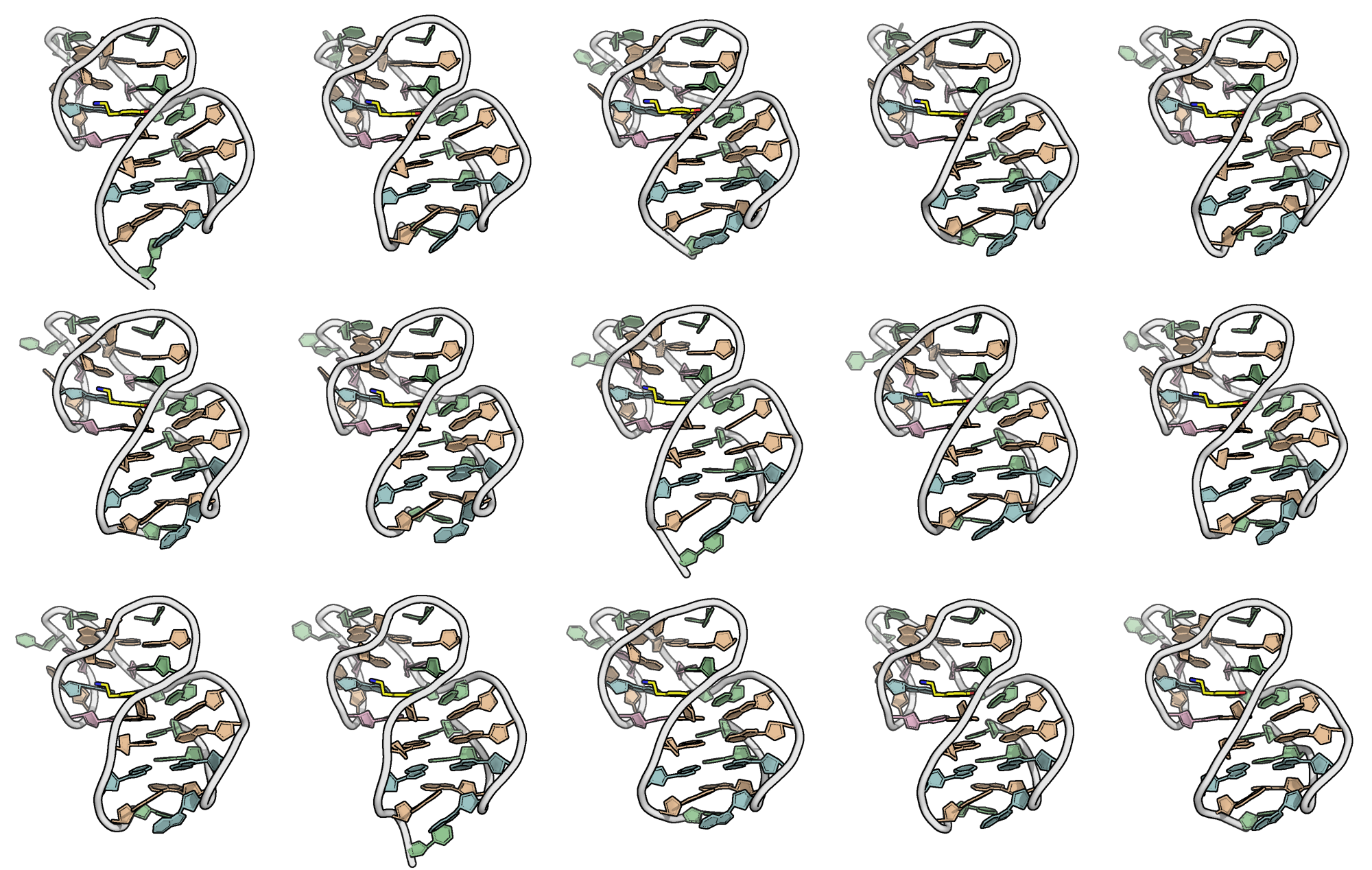
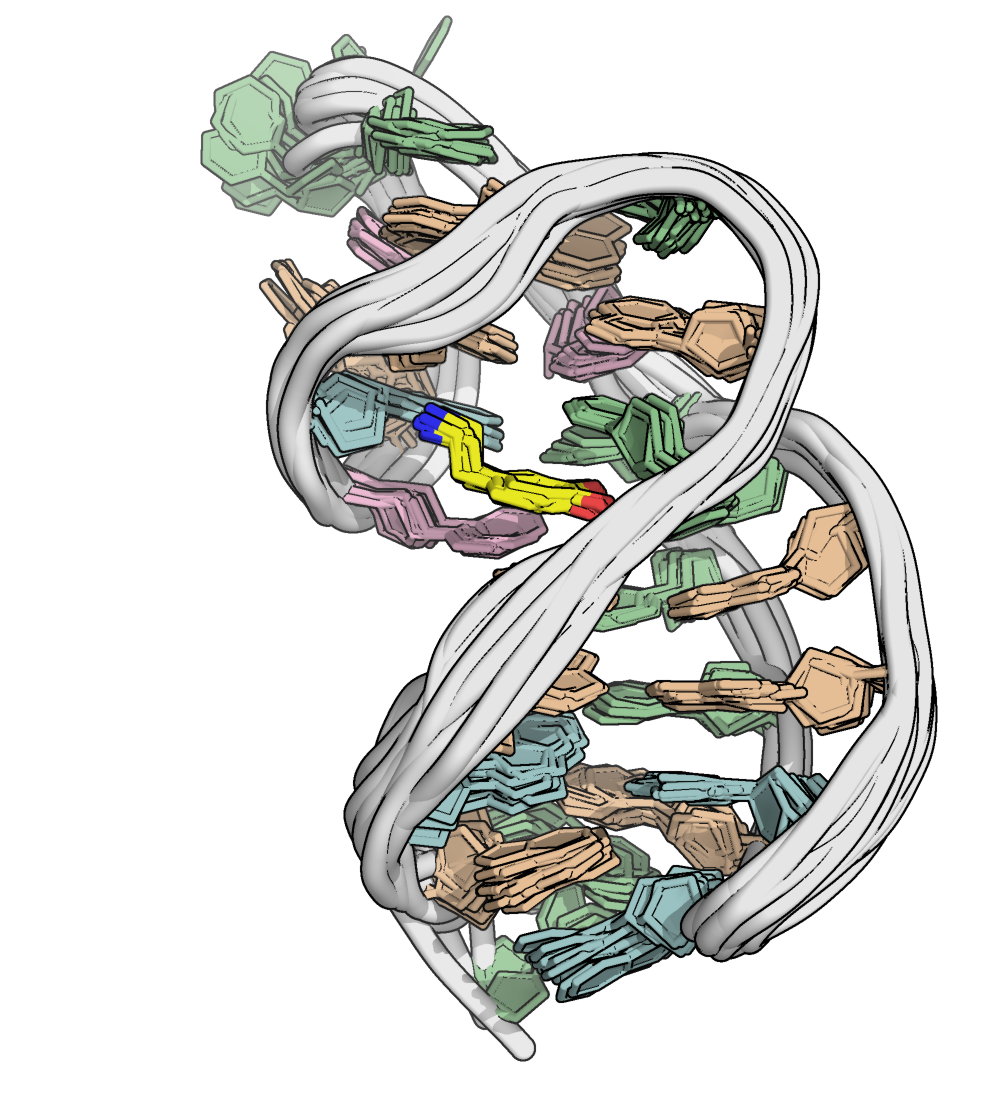
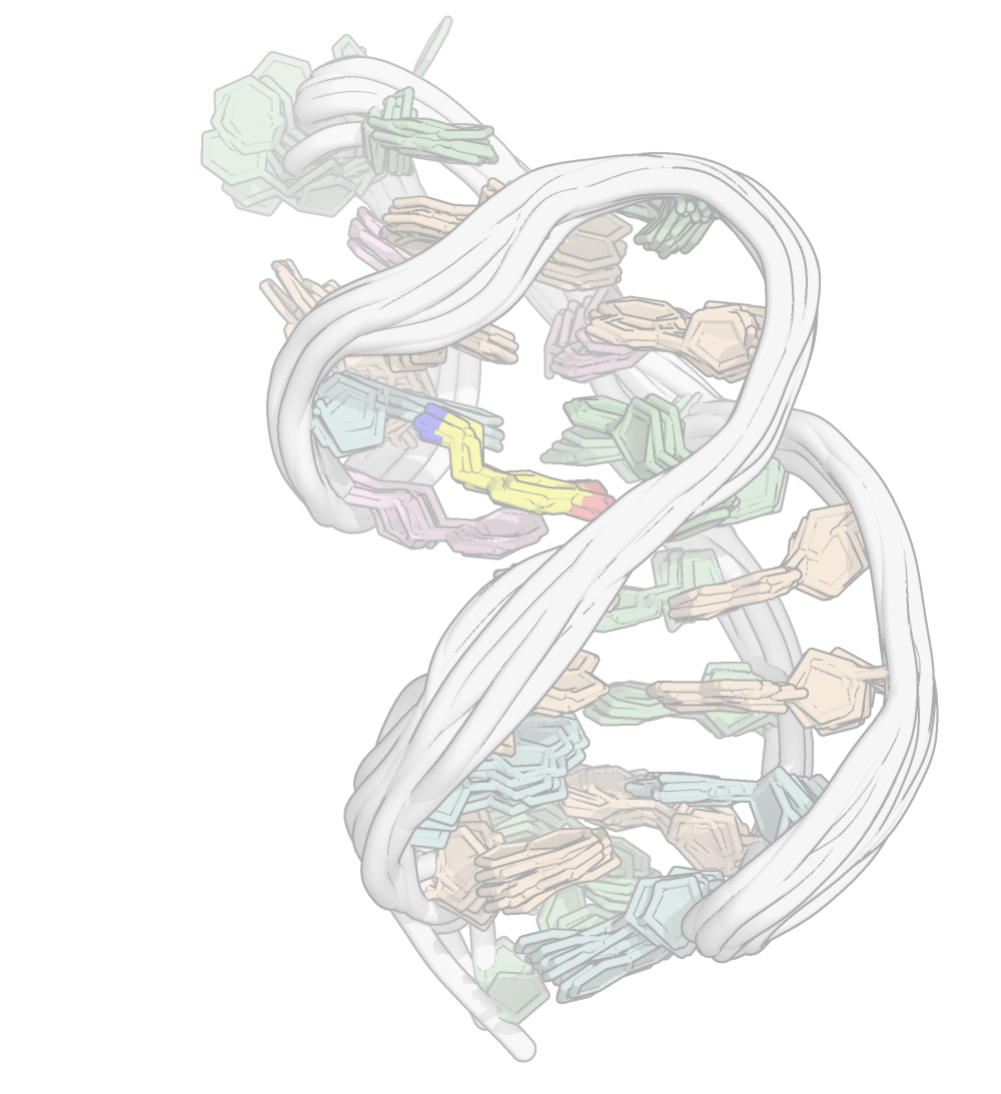

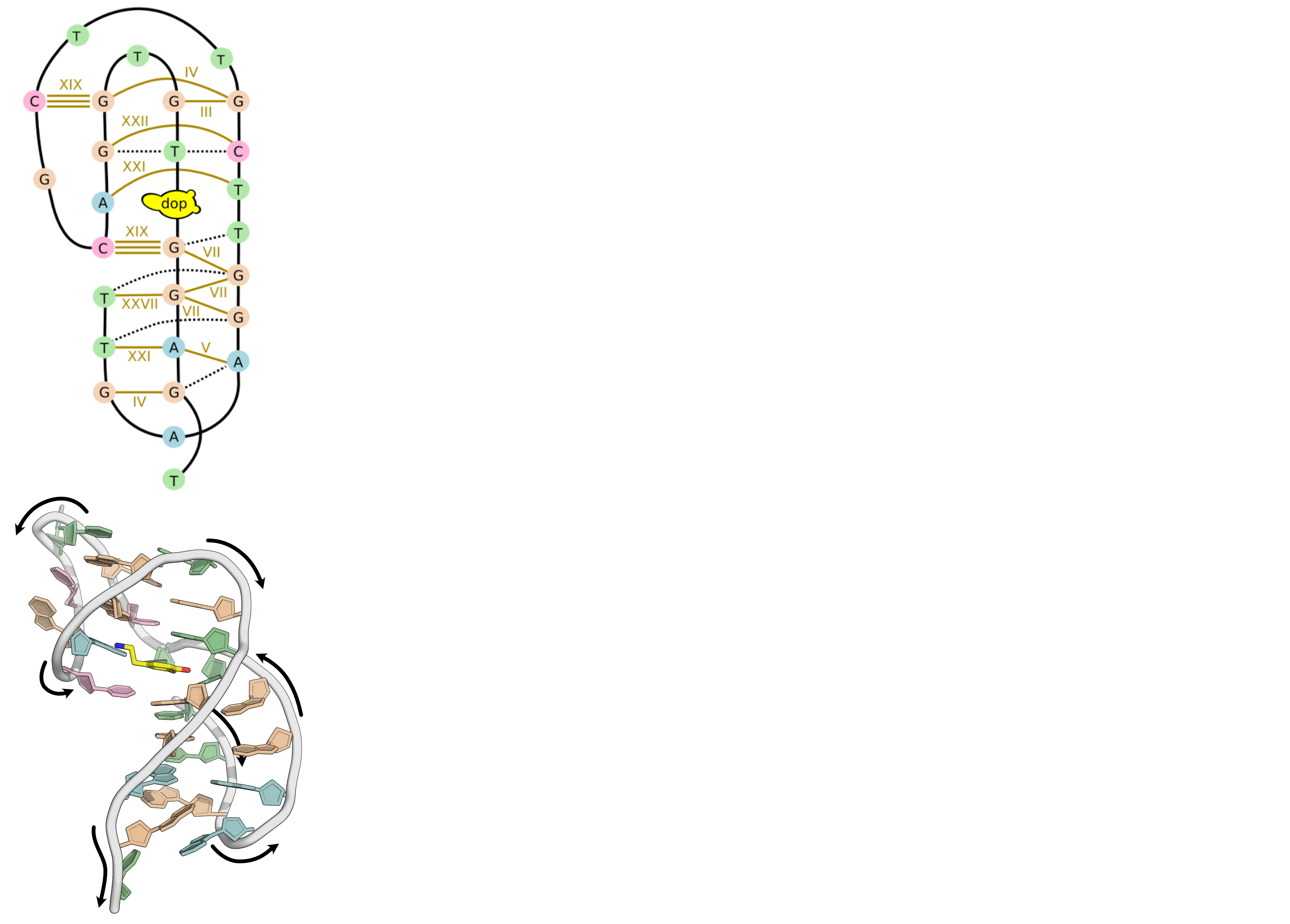
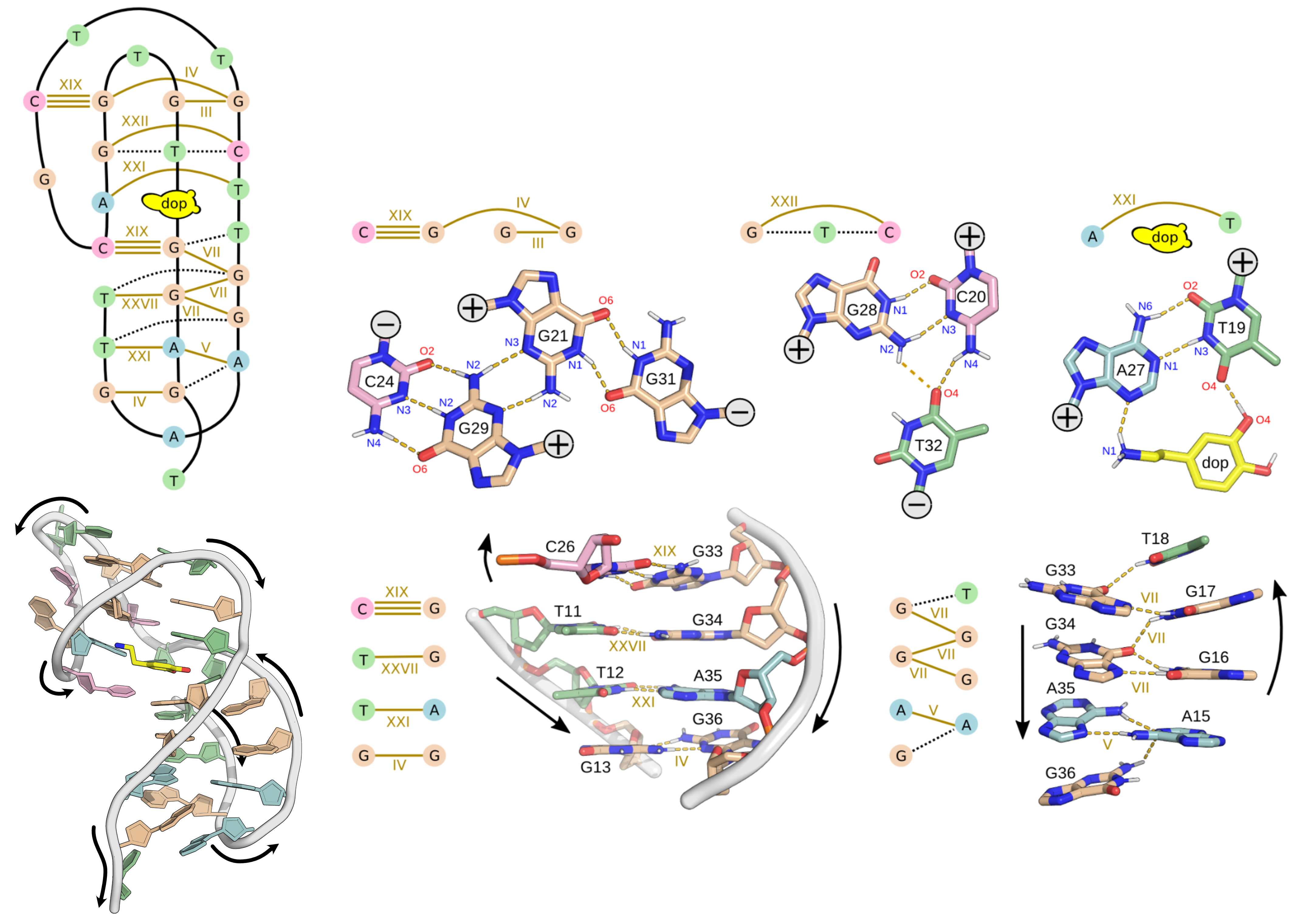
- CASP16
Critical Assessment of Techniques for Protein Structure Prediction- 107 models submitted
- 0 with RMSD < 10 Å
Funding sources


Thank you for your attention!